 Tweet
Tweet
A new pandemic influenza A(H1N1) genetic variant predominated in the winter 2010 influenza season in Australia, New Zealand and Singapore (Euro Surveill., edited)
Eurosurveillance, Volume 15, Issue 42, 21 October 2010
Research articles
A new pandemic influenza A(H1N1) genetic variant predominated in the winter 2010 influenza season in Australia, New Zealand and Singapore
I G Barr ( )<sup>1</sup>, L Cui<sup>2</sup>, N Komadina<sup>1</sup>, R T Lee<sup>3</sup>, R T Lin<sup>2</sup>, Y Deng<sup>1</sup>, N Caldwell<sup>1</sup>, R Shaw<sup>1</sup>, S Maurer-Stroh<sup>3</sup>
)<sup>1</sup>, L Cui<sup>2</sup>, N Komadina<sup>1</sup>, R T Lee<sup>3</sup>, R T Lin<sup>2</sup>, Y Deng<sup>1</sup>, N Caldwell<sup>1</sup>, R Shaw<sup>1</sup>, S Maurer-Stroh<sup>3</sup>
<hr>
Citation style for this article: Barr IG, Cui L, Komadina N, Lee RT, Lin RT, Deng Y, Caldwell N, Shaw R, Maurer-Stroh S. A new pandemic influenza A(H1N1) genetic variant predominated in the winter 2010 influenza season in Australia, New Zealand and Singapore. Euro Surveill. 2010;15(42):pii=19692. Available online: http://www.eurosurveillance.org/View...rticleId=19692
Date of submission: 14 September 2010
<hr>
Pandemic H1N1 influenza virus is of global health concern and is currently the predominant influenza virus subtype circulating in the southern hemisphere 2010 winter. The virus has changed little since it emerged in 2009, however, in this report we describe several genetically distinct changes in the pandemic H1N1 influenza virus. These variants were first detected in Singapore in early 2010 and have subsequently spread through Australia and New Zealand. At this stage, these signature changes in the haemagglutinin and neuraminidase proteins have not resulted in significant antigenic changes which might make the current vaccine less effective, but such adaptive mutations should be carefully monitored as the northern hemisphere approaches its winter influenza season.
<hr>
Since its emergence in early 2009 [1] the pandemic influenza A(H1N1) virus has remained closely related to one of the earliest viruses detected, A/California/7/2009, with little change in the viruses' genetic makeup in even the most variable genes, haemagglutinin (HA) and neuraminidase (NA). This lack of drift was reflected in the World Health Organization's (WHO) Vaccine formulation decisions which recommended an A/California/7/2009-like pandemic influenza A(H1N1) virus for both the southern hemisphere 2010 and the northern hemisphere 2010-11 influenza vaccines [2]. While some genetic variants have been reported such as the D222G (D239G numbering if starting at the first methionine) HA mutation which was linked with more severe outcomes following pandemic influenza virus infection [3] and a more commonly seen E391K change in the HA gene [4] during late 2009, no clear variant has predominated in a country or region and no vaccine update has been forthcoming. This report, however, describes the recent emergence in Singapore and subsequent spread of a genetic variant of the pandemic influenza A(H1N1) virus to Australia and New Zealand during their 2010 winter influenza season, where it now predominates and has been detected in some vaccine breakthroughs and fatal cases.
Genetic characterisation of the pandemic influenza A(H1N1) variant
We sequenced the HA, NA and other genes of 2010 pandemic influenza A(H1N1) viruses from Singapore, Australia, New Zealand and elsewhere using conventional Sanger sequencing. Viruses early in 2010 (January to April) from Singapore and Australia showed the E391K (numbering beginning at the first methionine in HA; equivalent to E374K if starting after the signal peptide sequence in HA at DTLC) change in the HA but were scattered throughout the phylogenetic trees for HA (Figure 1) and the whole genome (Figure 2).
Figure 1. Phylogenetic analysis of haemagglutinin sequences from recent pandemic influenza A(H1N1) viruses

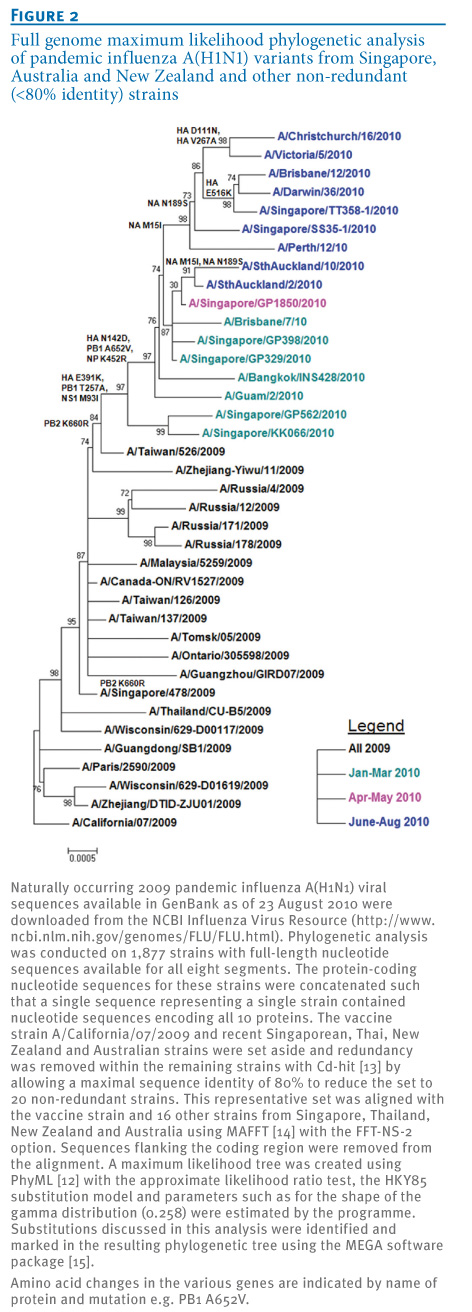
Figure 2. Full genome maximum likelihood phylogenetic analysis of pandemic influenza A(H1N1) variants from Singapore, Australia and New Zealand and other non-redundant (<80% identity) strains

On 13 April 2010 an influenza A(H1N1) strain, A/Singapore/CC01/2010, was detected in Singapore that had further changes in HA (N142D; numbering beginning at the first M in HA; equivalent to N125D if starting after the signal peptide sequence in HA at DTLC) and in NA (M15I, N189S). Viruses with these changes then increased in frequency during May and June 2010 in Singapore and became the predominant viruses by mid-2010. Of the pandemic influenza viruses sequenced in Australia in 2010, those sampled in January and February mostly had the E391K change.
Viruses with the dual HA mutation (E391K and N142D) were first detected in late April 2010 (e.g. A/Brisbane/10/2010, sample date 29 April 2010), and by June 2010, viruses with these HA (and NA) changes predominated. In the North and South Islands of New Zealand, viruses that were collected in July and August 2010 also showed this dual change in the HA along with the NA changes. Viruses with these genetic characteristics in the HA protein have only been detected sporadically in some other countries (e.g. Guam; Figure 1, Table 1), and the complete set of changes in HA and NA has not yet been reported in the northern hemisphere to date in 2010.
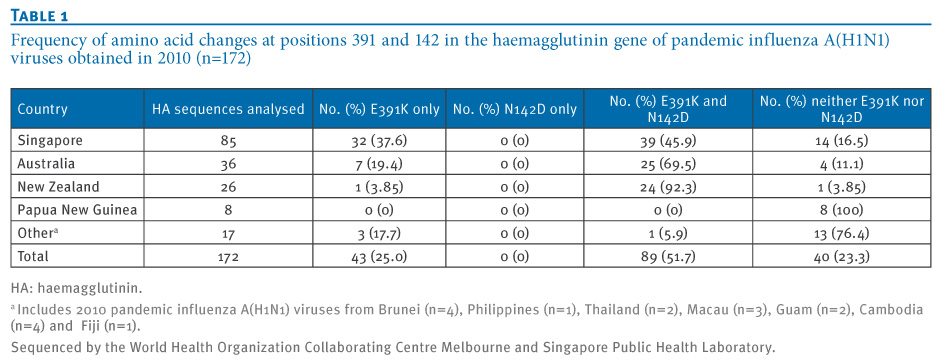
Table 1. Frequency of amino acid changes at positions 391 and 142 in the haemagglutinin gene of pandemic influenza A(H1N1) viruses obtained in 2010 (n=172)

These variant viruses have also been associated with several vaccine breakthroughs and a number of fatalities in both Singapore and Australia (labelled 'dec' in Figure 1). Examination of other gene segments of several pandemic influenza A(H1N1) variants showed that the other six segments were all very similar to the A/California/7/2009 strain (nucleotide identity ranged from >99% to 100%) with no evidence of gene reassorting between the pandemic influenza (H1N1) virus and seasonal influenza A(H1N1) or H3N2 viruses or another influenza A subtype. Nevertheless, as marked in the whole genome phylogenetic tree (Figure 2), some additional mutations in the other gene segments (PB2, PB1, NP, NS1) appeared commonly among the recent variant strains, but the significance of these changes remains to be determined.
To further investigate the importance of these surface antigen mutations, we built a structural homology model of HA from the A/Brisbane/10/2010(H1N1) virus based on the template structure of A/California/04/2009(H1N1) (PDB:3LZG) [5] using MODELLER with loop refinement [6] and ProQ [7] for model quality control. In Figure 3, we superimpose our model with the complex of the antigenically similar HA of the 1918 influenza A(H1) virus bound to an antibody that recognises the classical Sa epitope (PDB:3LZF) [5]. We show that N142D is centrally located in this epitope, which led us to further investigate the effect of the mutation on antigenic properties with haemagglutination inhibition assays.
Figure 3. Structural model of influenza A/Brisbane/10/2010(H1N1) haemagglutinin

Adding to the possibility of the N142D mutation affecting antigenicity, the equivalent mutation N129D(H3)/N124D(H5) in influenza A/Mallard/Pennsylvania/10218/84(H5N2) virus was previously reported to cause antigenic drift as an escape mutant [8]. However, the findings in the context of avian H5 may not be easily transferable to the swine-origin H1. Generally, a single mutation will only partially affect antigenicity as typically several mutations in the same epitope are needed to seriously alter vaccine efficacy.
An additional mutation in the HA sequence, D111N, was common among samples from New Zealand, and an equivalent mutation in avian influenza has been reported to be related to a shift in host specificity (from avian towards human) which could hypothetically mean a small fitness advantage in the human host [9]. The equivalent mutation (referred to as D94N in [9]) enhanced binding of HA to the human-type SA--2,6-Gal receptor and decreased binding to the avian-type SA--2,3-Gal receptor. It was also observed that the mutation was able to enhance HA-mediated membrane fusion in mammalian cells. Structurally, D111N is located on the outside of the bottom of the sialic acid binding pocket with the side chain pointing to the outside (Figure 3) and the mechanism that causes the reported effect is not fully clear.
HA D111N is almost exclusively found in combination with another mutation, HA V267A, which is located at an internal beta sheet below the receptor binding pocket facing the Sa epitope (Figure 2). Exchanging valine for the smaller alanine at this position creates a small cavity which may slightly alter the surface of the epitope on top and could add to the effects of N142D. However, so far the HA D111N and V267A mutations have only occurred in a close temporal and geographic context (four in New Zealand and three in eastern Australia in July and August 2010, see Figure 1) and their increased local occurrence may simply be due to founder effects.
Two additional mutations occurred in the NA sequence, M15I and N189S, which were predominant in viruses from Singapore and Australia by mid-2010. NA M15I is located in the signal peptide region. The signal peptide is the motif required for cell surface expression of the viral protein and its existence and quality can be predicted with the programme SignalP 3.0 [10]. For the NA M15I mutation, the prediction score (D-score) increases from 0.326 for M15 to 0.404 for I15. This could hypothetically indicate that the mutated version represents a better signal peptide with potentially increasing secretion and surface expression efficiency, but this needs to be further tested experimentally. NA N189S on the other hand is located at the bottom side of the NA structure, far away from the sialic acid- and drug-binding pocket and any phenotypic change cannot easily be predicted.
Antigenic analysis of variant viruses
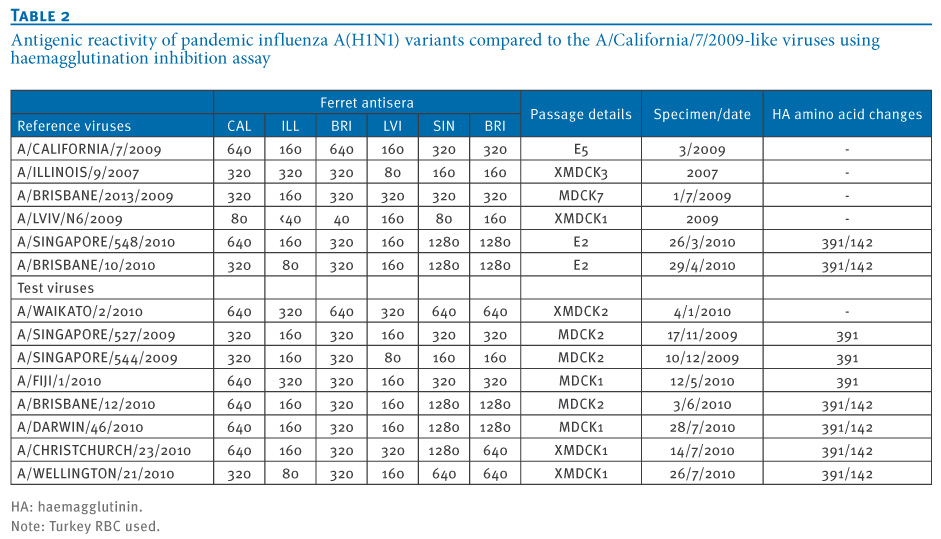
While genetic differences were apparent in this variant group of pandemic influenza A(H1N1) viruses, when they were assessed for antigenic variation in haemagglutination inhibition assays (HI) using ferret antisera raised to A/California/7/2009-like viruses and viruses from the new variant group (e.g. A/Singapore/548/2010, A/Brisbane10/2010), no differences in titres were apparent, indicating that these viruses were not antigenically distinguishable from the reference and vaccine virus A/California/7/2009 (Table 2).
Table 2. Antigenic reactivity of pandemic influenza A(H1N1) variants compared to the A/California/7/2009-like viruses using haemagglutination inhibition assay

Further antigenic analysis was performed using a small human serum panel (n=48) containing pre- and post-vaccination sera from Australian adults (24 subjects between the age of 18 and 59 years) and elderly subjects (24 subjects over the age of 60 years) who were given a single dose of inactivated 2010 Australian seasonal influenza vaccine (CSL Fluvax; CSL Limited, Australia) which contained an A/California/7/2009-like pandemic influenza A(H1N1) virus, an A/Perth/16/2009-like A(H3N2) virus and a B/Brisbane/60/2008-like B virus. The geometric mean HI titre (GMT) in the sera of all vaccinated subjects was reduced by 53% when tested against an egg-grown A/Brisbane/10/2010 virus (one of the genetically variant viruses) compared to the GMT obtained against egg-grown wildtype A/California/7/2009 virus. Despite some reduction in HI titres with human post-vaccination sera, there were no clear differences with ferret sera, suggesting that there are no major antigenic differences in these variant viruses at this stage in their evolution and that they still share most of their antigenic properties with the early pandemic influenza A(H1N1) viruses.
Discussion
While the 2009 pandemic has recently been downgraded by the WHO [11], the pandemic influenza A(H1N1) virus still remains the predominant influenza virus in most countries including those in the southern hemisphere that recently experienced their winter influenza season (with the exception of South Africa where influenza B and A(H3N2) viruses have predominated in 2010) [11]. To date there has been little change detected in either the genetic or antigenic characteristics of the pandemic H1N1 influenza virus in the nearly 18 months that it has infected humans. No clear variant has appeared apart from minor changes occurring in the HA, NA and other viral genes during this time. Recently however a genetically distinct variant containing several signature amino acid changes in both the HA and NA genes has emerged in Singapore, Australia and New Zealand, coinciding with the winter influenza season in the latter two countries. While the combination of HA mutations at E391K and N142D has been seen sporadically in isolates in Korea and the United States in November 2009 (ADM21270, ADM21278, ADL59660, ADD74728), the first appearance of the double HA and double NA change has been in April in Singapore (A/Singapore/CC01/2010). Similar viruses have also been detected in Guam (A/Guam/2/2010(H1N1)) and Thailand (A/Bangkok/INS428/2010(H1N1)) in March 2010; they were lacking the NA mutations but at least partially shared the changes in other segments (PB2 K660R, NS1 M93I, PB1 T257A and A652V, NP K452R) which makes these strains closely related to A/Singapore/GP329/2010 which was isolated in January 2010. Strains with the full set of characteristic mutations of the new variant have not yet been reported in other regions and have not appeared in genetic databases to date.
It remains to be seen whether this variant will continue to predominate for the rest of the influenza season in Oceania and in other parts of the southern hemisphere and then spread to the northern hemisphere or merely die out. Already this variant virus has been associated with several vaccine breakthroughs in teenagers and adults vaccinated in 2010 with monovalent pandemic influenza vaccine as well as a number of fatal cases from whom the variant virus was isolated. Unfortunately we did not have access to the comprehensive patient records that may have enabled us to determine the relative frequency of vaccine breakthroughs with this variant compared to the non-variant. This information is important to eliminate other confounding factors such as the age of the vaccinee, time since they were vaccinated or if they were taking immuosuppressive drugs, all of which might impact on their level of protection following vaccination. It is therefore not known at this time if the amino acid changes in this variant virus are responsible for these vaccine breakthroughs or deaths, or if they are simply a result of this virus genotype being the predominant virus in circulation during this period. The HA and NA amino acid changes seen in the variant are present both in the original clinical samples and in viruses isolated in MDCK cells and are retained in viruses isolated directly in embryonated hens eggs. Careful studies are underway to determine if the variant viruses grow better than other influenza A/California/7/2009-like viruses but preliminary data indicate that these variant viruses grow as well or better than viruses without these characteristic amino acid changes both in MDCK cell culture and in embryonated hens eggs (data not shown) and this may make them useful vaccine virus seeds. If confirmed this may also explain the rapid spread and predominance of this variant virus. Further animal and human transmission and growth studies will be required to support this initial finding.
Conclusions
A new genetic variant of the pandemic influenza (H1N1) virus has emerged in Singapore, Australia and New Zealand in the second and third quarters of 2010 that does not appear at this stage to represent a significant antigenic change for the virus. However, it may represent the start of more dramatic antigenic drift of the pandemic influenza A(H1N1) viruses that may require a vaccine update sooner than might have been expected, with a new human influenza virus.
Acknowledgements
The authors would like to thank the WHO National Influenza Centres and other laboratories for supplying viruses used in this study, Chua Sin Ying for help with the Bioinformatics analysis and Shiau Pheng Phuah, Shu Meng, Meng Li Teo for their technical support. The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health and Ageing.
<hr>
References
-
------
Eurosurveillance, Volume 15, Issue 42, 21 October 2010
Research articles
A new pandemic influenza A(H1N1) genetic variant predominated in the winter 2010 influenza season in Australia, New Zealand and Singapore
I G Barr (
- World Health Organization Collaborating Centre for Reference and Research on Influenza, Melbourne, Australia
- National Public Health Laboratory, Ministry of Health, Singapore
- Bioinformatics Institute (BII), A*STAR, Singapore
<hr>
Citation style for this article: Barr IG, Cui L, Komadina N, Lee RT, Lin RT, Deng Y, Caldwell N, Shaw R, Maurer-Stroh S. A new pandemic influenza A(H1N1) genetic variant predominated in the winter 2010 influenza season in Australia, New Zealand and Singapore. Euro Surveill. 2010;15(42):pii=19692. Available online: http://www.eurosurveillance.org/View...rticleId=19692
Date of submission: 14 September 2010
<hr>
Pandemic H1N1 influenza virus is of global health concern and is currently the predominant influenza virus subtype circulating in the southern hemisphere 2010 winter. The virus has changed little since it emerged in 2009, however, in this report we describe several genetically distinct changes in the pandemic H1N1 influenza virus. These variants were first detected in Singapore in early 2010 and have subsequently spread through Australia and New Zealand. At this stage, these signature changes in the haemagglutinin and neuraminidase proteins have not resulted in significant antigenic changes which might make the current vaccine less effective, but such adaptive mutations should be carefully monitored as the northern hemisphere approaches its winter influenza season.
<hr>
Since its emergence in early 2009 [1] the pandemic influenza A(H1N1) virus has remained closely related to one of the earliest viruses detected, A/California/7/2009, with little change in the viruses' genetic makeup in even the most variable genes, haemagglutinin (HA) and neuraminidase (NA). This lack of drift was reflected in the World Health Organization's (WHO) Vaccine formulation decisions which recommended an A/California/7/2009-like pandemic influenza A(H1N1) virus for both the southern hemisphere 2010 and the northern hemisphere 2010-11 influenza vaccines [2]. While some genetic variants have been reported such as the D222G (D239G numbering if starting at the first methionine) HA mutation which was linked with more severe outcomes following pandemic influenza virus infection [3] and a more commonly seen E391K change in the HA gene [4] during late 2009, no clear variant has predominated in a country or region and no vaccine update has been forthcoming. This report, however, describes the recent emergence in Singapore and subsequent spread of a genetic variant of the pandemic influenza A(H1N1) virus to Australia and New Zealand during their 2010 winter influenza season, where it now predominates and has been detected in some vaccine breakthroughs and fatal cases.
Genetic characterisation of the pandemic influenza A(H1N1) variant
We sequenced the HA, NA and other genes of 2010 pandemic influenza A(H1N1) viruses from Singapore, Australia, New Zealand and elsewhere using conventional Sanger sequencing. Viruses early in 2010 (January to April) from Singapore and Australia showed the E391K (numbering beginning at the first methionine in HA; equivalent to E374K if starting after the signal peptide sequence in HA at DTLC) change in the HA but were scattered throughout the phylogenetic trees for HA (Figure 1) and the whole genome (Figure 2).
Figure 1. Phylogenetic analysis of haemagglutinin sequences from recent pandemic influenza A(H1N1) viruses
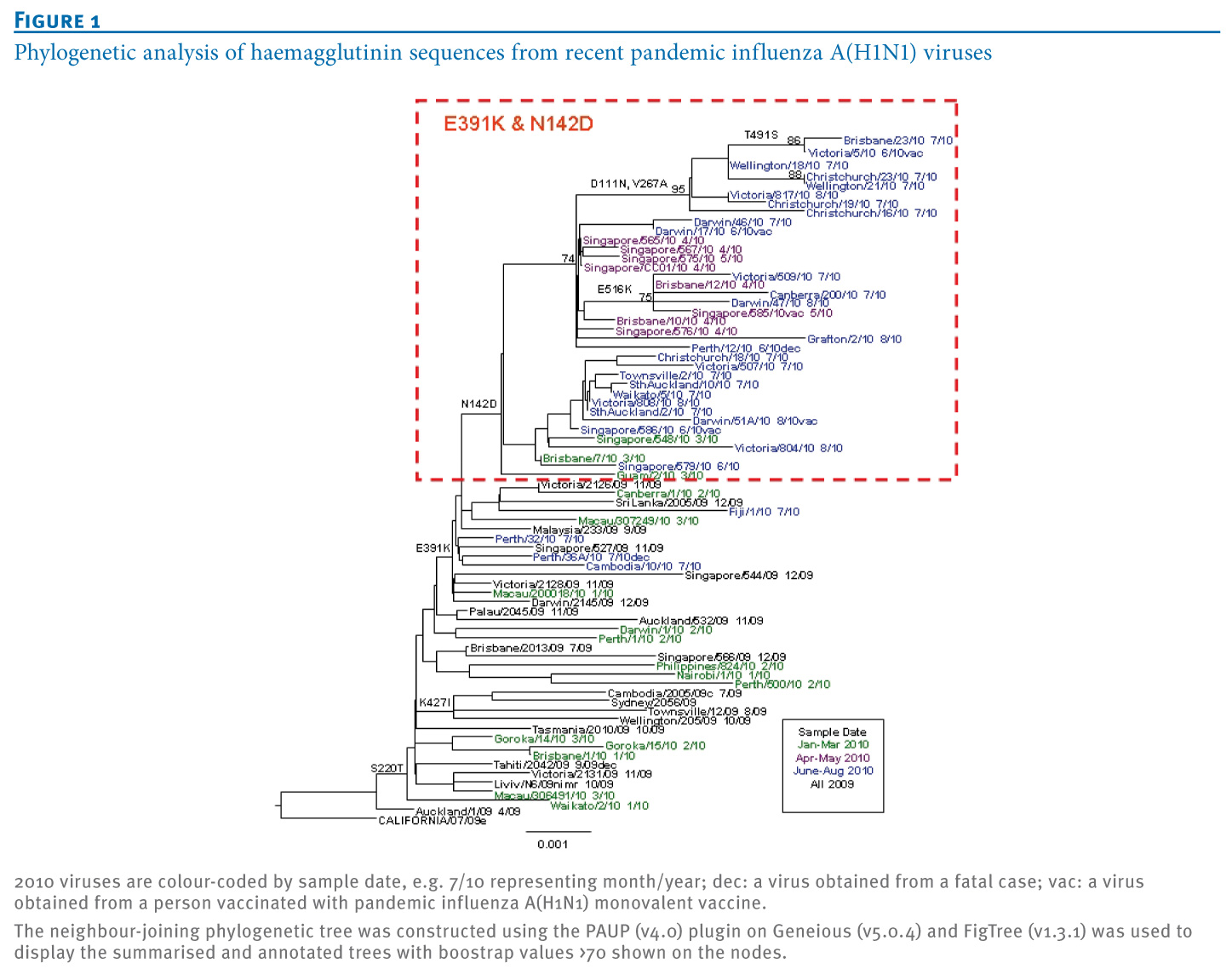
Figure 2. Full genome maximum likelihood phylogenetic analysis of pandemic influenza A(H1N1) variants from Singapore, Australia and New Zealand and other non-redundant (<80% identity) strains
On 13 April 2010 an influenza A(H1N1) strain, A/Singapore/CC01/2010, was detected in Singapore that had further changes in HA (N142D; numbering beginning at the first M in HA; equivalent to N125D if starting after the signal peptide sequence in HA at DTLC) and in NA (M15I, N189S). Viruses with these changes then increased in frequency during May and June 2010 in Singapore and became the predominant viruses by mid-2010. Of the pandemic influenza viruses sequenced in Australia in 2010, those sampled in January and February mostly had the E391K change.
Viruses with the dual HA mutation (E391K and N142D) were first detected in late April 2010 (e.g. A/Brisbane/10/2010, sample date 29 April 2010), and by June 2010, viruses with these HA (and NA) changes predominated. In the North and South Islands of New Zealand, viruses that were collected in July and August 2010 also showed this dual change in the HA along with the NA changes. Viruses with these genetic characteristics in the HA protein have only been detected sporadically in some other countries (e.g. Guam; Figure 1, Table 1), and the complete set of changes in HA and NA has not yet been reported in the northern hemisphere to date in 2010.
Table 1. Frequency of amino acid changes at positions 391 and 142 in the haemagglutinin gene of pandemic influenza A(H1N1) viruses obtained in 2010 (n=172)
These variant viruses have also been associated with several vaccine breakthroughs and a number of fatalities in both Singapore and Australia (labelled 'dec' in Figure 1). Examination of other gene segments of several pandemic influenza A(H1N1) variants showed that the other six segments were all very similar to the A/California/7/2009 strain (nucleotide identity ranged from >99% to 100%) with no evidence of gene reassorting between the pandemic influenza (H1N1) virus and seasonal influenza A(H1N1) or H3N2 viruses or another influenza A subtype. Nevertheless, as marked in the whole genome phylogenetic tree (Figure 2), some additional mutations in the other gene segments (PB2, PB1, NP, NS1) appeared commonly among the recent variant strains, but the significance of these changes remains to be determined.
To further investigate the importance of these surface antigen mutations, we built a structural homology model of HA from the A/Brisbane/10/2010(H1N1) virus based on the template structure of A/California/04/2009(H1N1) (PDB:3LZG) [5] using MODELLER with loop refinement [6] and ProQ [7] for model quality control. In Figure 3, we superimpose our model with the complex of the antigenically similar HA of the 1918 influenza A(H1) virus bound to an antibody that recognises the classical Sa epitope (PDB:3LZF) [5]. We show that N142D is centrally located in this epitope, which led us to further investigate the effect of the mutation on antigenic properties with haemagglutination inhibition assays.
Figure 3. Structural model of influenza A/Brisbane/10/2010(H1N1) haemagglutinin
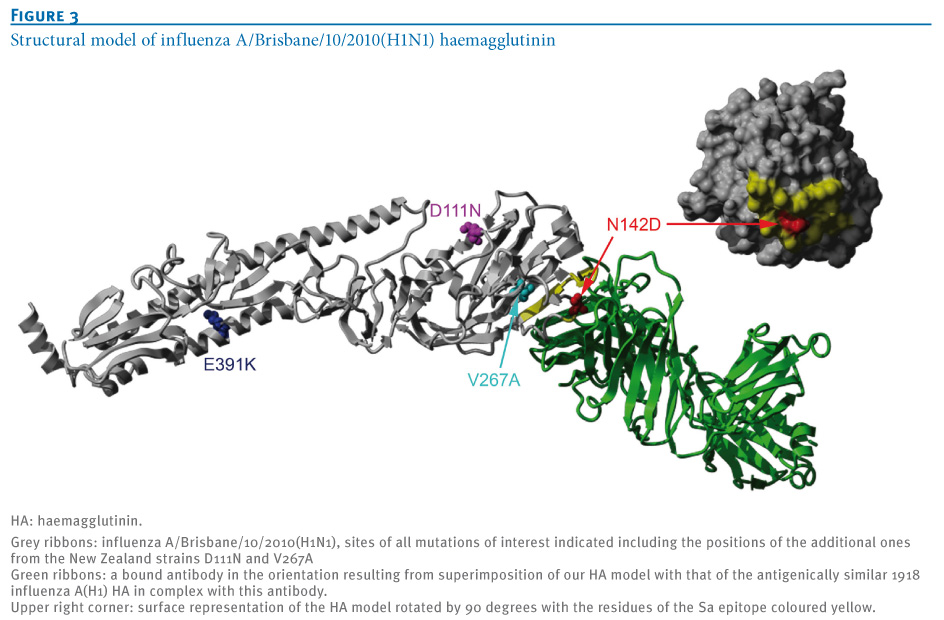
Adding to the possibility of the N142D mutation affecting antigenicity, the equivalent mutation N129D(H3)/N124D(H5) in influenza A/Mallard/Pennsylvania/10218/84(H5N2) virus was previously reported to cause antigenic drift as an escape mutant [8]. However, the findings in the context of avian H5 may not be easily transferable to the swine-origin H1. Generally, a single mutation will only partially affect antigenicity as typically several mutations in the same epitope are needed to seriously alter vaccine efficacy.
An additional mutation in the HA sequence, D111N, was common among samples from New Zealand, and an equivalent mutation in avian influenza has been reported to be related to a shift in host specificity (from avian towards human) which could hypothetically mean a small fitness advantage in the human host [9]. The equivalent mutation (referred to as D94N in [9]) enhanced binding of HA to the human-type SA--2,6-Gal receptor and decreased binding to the avian-type SA--2,3-Gal receptor. It was also observed that the mutation was able to enhance HA-mediated membrane fusion in mammalian cells. Structurally, D111N is located on the outside of the bottom of the sialic acid binding pocket with the side chain pointing to the outside (Figure 3) and the mechanism that causes the reported effect is not fully clear.
HA D111N is almost exclusively found in combination with another mutation, HA V267A, which is located at an internal beta sheet below the receptor binding pocket facing the Sa epitope (Figure 2). Exchanging valine for the smaller alanine at this position creates a small cavity which may slightly alter the surface of the epitope on top and could add to the effects of N142D. However, so far the HA D111N and V267A mutations have only occurred in a close temporal and geographic context (four in New Zealand and three in eastern Australia in July and August 2010, see Figure 1) and their increased local occurrence may simply be due to founder effects.
Two additional mutations occurred in the NA sequence, M15I and N189S, which were predominant in viruses from Singapore and Australia by mid-2010. NA M15I is located in the signal peptide region. The signal peptide is the motif required for cell surface expression of the viral protein and its existence and quality can be predicted with the programme SignalP 3.0 [10]. For the NA M15I mutation, the prediction score (D-score) increases from 0.326 for M15 to 0.404 for I15. This could hypothetically indicate that the mutated version represents a better signal peptide with potentially increasing secretion and surface expression efficiency, but this needs to be further tested experimentally. NA N189S on the other hand is located at the bottom side of the NA structure, far away from the sialic acid- and drug-binding pocket and any phenotypic change cannot easily be predicted.
Antigenic analysis of variant viruses
While genetic differences were apparent in this variant group of pandemic influenza A(H1N1) viruses, when they were assessed for antigenic variation in haemagglutination inhibition assays (HI) using ferret antisera raised to A/California/7/2009-like viruses and viruses from the new variant group (e.g. A/Singapore/548/2010, A/Brisbane10/2010), no differences in titres were apparent, indicating that these viruses were not antigenically distinguishable from the reference and vaccine virus A/California/7/2009 (Table 2).
Table 2. Antigenic reactivity of pandemic influenza A(H1N1) variants compared to the A/California/7/2009-like viruses using haemagglutination inhibition assay
Further antigenic analysis was performed using a small human serum panel (n=48) containing pre- and post-vaccination sera from Australian adults (24 subjects between the age of 18 and 59 years) and elderly subjects (24 subjects over the age of 60 years) who were given a single dose of inactivated 2010 Australian seasonal influenza vaccine (CSL Fluvax; CSL Limited, Australia) which contained an A/California/7/2009-like pandemic influenza A(H1N1) virus, an A/Perth/16/2009-like A(H3N2) virus and a B/Brisbane/60/2008-like B virus. The geometric mean HI titre (GMT) in the sera of all vaccinated subjects was reduced by 53% when tested against an egg-grown A/Brisbane/10/2010 virus (one of the genetically variant viruses) compared to the GMT obtained against egg-grown wildtype A/California/7/2009 virus. Despite some reduction in HI titres with human post-vaccination sera, there were no clear differences with ferret sera, suggesting that there are no major antigenic differences in these variant viruses at this stage in their evolution and that they still share most of their antigenic properties with the early pandemic influenza A(H1N1) viruses.
Discussion
While the 2009 pandemic has recently been downgraded by the WHO [11], the pandemic influenza A(H1N1) virus still remains the predominant influenza virus in most countries including those in the southern hemisphere that recently experienced their winter influenza season (with the exception of South Africa where influenza B and A(H3N2) viruses have predominated in 2010) [11]. To date there has been little change detected in either the genetic or antigenic characteristics of the pandemic H1N1 influenza virus in the nearly 18 months that it has infected humans. No clear variant has appeared apart from minor changes occurring in the HA, NA and other viral genes during this time. Recently however a genetically distinct variant containing several signature amino acid changes in both the HA and NA genes has emerged in Singapore, Australia and New Zealand, coinciding with the winter influenza season in the latter two countries. While the combination of HA mutations at E391K and N142D has been seen sporadically in isolates in Korea and the United States in November 2009 (ADM21270, ADM21278, ADL59660, ADD74728), the first appearance of the double HA and double NA change has been in April in Singapore (A/Singapore/CC01/2010). Similar viruses have also been detected in Guam (A/Guam/2/2010(H1N1)) and Thailand (A/Bangkok/INS428/2010(H1N1)) in March 2010; they were lacking the NA mutations but at least partially shared the changes in other segments (PB2 K660R, NS1 M93I, PB1 T257A and A652V, NP K452R) which makes these strains closely related to A/Singapore/GP329/2010 which was isolated in January 2010. Strains with the full set of characteristic mutations of the new variant have not yet been reported in other regions and have not appeared in genetic databases to date.
It remains to be seen whether this variant will continue to predominate for the rest of the influenza season in Oceania and in other parts of the southern hemisphere and then spread to the northern hemisphere or merely die out. Already this variant virus has been associated with several vaccine breakthroughs in teenagers and adults vaccinated in 2010 with monovalent pandemic influenza vaccine as well as a number of fatal cases from whom the variant virus was isolated. Unfortunately we did not have access to the comprehensive patient records that may have enabled us to determine the relative frequency of vaccine breakthroughs with this variant compared to the non-variant. This information is important to eliminate other confounding factors such as the age of the vaccinee, time since they were vaccinated or if they were taking immuosuppressive drugs, all of which might impact on their level of protection following vaccination. It is therefore not known at this time if the amino acid changes in this variant virus are responsible for these vaccine breakthroughs or deaths, or if they are simply a result of this virus genotype being the predominant virus in circulation during this period. The HA and NA amino acid changes seen in the variant are present both in the original clinical samples and in viruses isolated in MDCK cells and are retained in viruses isolated directly in embryonated hens eggs. Careful studies are underway to determine if the variant viruses grow better than other influenza A/California/7/2009-like viruses but preliminary data indicate that these variant viruses grow as well or better than viruses without these characteristic amino acid changes both in MDCK cell culture and in embryonated hens eggs (data not shown) and this may make them useful vaccine virus seeds. If confirmed this may also explain the rapid spread and predominance of this variant virus. Further animal and human transmission and growth studies will be required to support this initial finding.
Conclusions
A new genetic variant of the pandemic influenza (H1N1) virus has emerged in Singapore, Australia and New Zealand in the second and third quarters of 2010 that does not appear at this stage to represent a significant antigenic change for the virus. However, it may represent the start of more dramatic antigenic drift of the pandemic influenza A(H1N1) viruses that may require a vaccine update sooner than might have been expected, with a new human influenza virus.
Acknowledgements
The authors would like to thank the WHO National Influenza Centres and other laboratories for supplying viruses used in this study, Chua Sin Ying for help with the Bioinformatics analysis and Shiau Pheng Phuah, Shu Meng, Meng Li Teo for their technical support. The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health and Ageing.
<hr>
References
- Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team, Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, et al. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med. 2009;360(25):2605-15.
- World Health Organization (WHO). Recommendations for influenza vaccines. Geneva:WHO; [Accessed 23 Aug 2010]. Available from: http://www.who.int/csr/disease/influ.../en/index.html
- Kilander A, Rykkvin R, Dudman SG, Hungnes O. Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome, Norway 2009-2010. Euro Surveill. 2010;15(9):pii=19498. Available from: http://www.eurosurveillance.org/View...rticleId=19498
- Maurer-Stroh S, Lee RT, Eisenhaber F, Cui L, Phuah SP, Lin RT. A new common mutation in the hemagglutinin of the 2009 (H1N1) influenza A virus. PLoS Curr. 2010 Jun 1:RRN1162.
- Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE Jr, Wilson IA. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science. 2010;328(5976):357-60.
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007 Nov;Chapter 2:Unit 2.9.
- Wallner B, Elofsson A. Can correct protein models be identified? Protein Sci. 2003;12(5):1073-86.
- Kaverin NV, Rudneva IA, Govorkova EA, Timofeeva TA, Shilov AA, Kochergin-Nikitsky KS, et al. Epitope mapping of the hemagglutinin molecule of a highly pathogenic H5N1 influenza virus by using monoclonal antibodies. J Virol. 2007; 81(23):12911-17.
- Su Y. Yang HY, Zhang BJ, Jia HL, Tien P. Analysis of a point mutation in H5N1 avian influenza virus haemagglutinin in relation to virus entry into live mammalian cells. Arch Virol. 2008;153(12): 2253-61.
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340(4):783-95.
- Nicoll A, Sprenger M. The end of the pandemic ? what will be the pattern of influenza in the 2010-11 European winter and beyond? . Euro Surveill. 2010;15(32):pii=19637. Available from: http://www.eurosurveillance.org/View...rticleId=19637
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696-704.
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658-9.
- Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33(2):511-18.
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol Biol Evol. 2007;24(8):1596-9.
-
------