 Tweet
Tweet
[Source: Eurosurveillance, full page: (LINK). Edited.]
Eurosurveillance, Volume 19, Issue 6, 13 February 2014
Rapid communications
Possible pandemic threat from new reassortment of influenza A(H7N9) virus in China
Z Meng<SUP>1</SUP>, R Han<SUP>2</SUP>, Y Hu<SUP>1</SUP>, Z Yuan<SUP>1</SUP>, S Jiang<SUP>1</SUP>, X Zhang<SUP>1</SUP><SUP>,3</SUP>, J Xu <SUP>1</SUP><SUP>,3</SUP>
<SUP></SUP>
<SUP>1</SUP>Shanghai Public Health Clinical Center and Institutes of Biomedical Sciences, Key Laboratory of Medical Molecular Virology of Ministry of Education/Health, Shanghai Medical College, Fudan University, Shanghai, China - 2 Department of Cell and Molecular Physiology, Loyola University Chicago Health Sciences Division, Maywood, IL, United States - 3 State Key Laboratory for Infectious Disease Prevention and Control, China Center for Disease Control and Prevention, Beijing, China
_____
Citation style for this article: Meng Z, Han R, Hu Y, Yuan Z, Jiang S, Zhang X, Xu J. Possible pandemic threat from new reassortment of influenza A(H7N9) virus in China. Euro Surveill. 2014;19(6):pii=20699. Available online: http://www.eurosurveillance.org/View...rticleId=20699
Date of submission: 29 January 2014
_____
Avian influenza A(H7N9) virus re-emerged in China in December 2013, after a decrease in the number of new cases during the preceding six months. Reassortment between influenza A(H7N9) and local H9N2 strains has spread from China's south-east coast to other regions. Three new reassortments of A(H7N9) virus were identified by phylogenetic analysis: between A(H7N9) and Zhejiang-derived strains, Guangdong/Hong Kong-derived strains or Hunan-derived A(H9N2) strains. Our findings suggest there is a possible risk that a pandemic could develop.
________
Recent re-emerged influenza A(H7N9) virus infections in China ? especially the rapid outbreak in Zhejiang province in December 2013, involving 60 cases [1] ? have raised concerns. Although several reports described the genetic characteristics of the virus [2-4], little is known about its further evolution after the initial outbreak in March 2013 [2] and the current re-emergence. As of 31 January 2014, there were a total of 260 cases: 127 of these have occurred in 2014 [5,6]. Cases have been reported from Zhejiang, Guangdong and Jiangsu provinces, Shanghai metropolitan area and Hong Kong in 2014 [6].
It is important to know whether new variants or lineages of influenza A(H7N9) virus are responsible for this re-emergence of the virus. In this study, four lineages and three new reassortments of A(H7N9) virus were identified by phylogenetic analysis and DNA mutation analysis of the PB1 gene.
Sequences analysis of PB1 genes from influenza A(H7N9) virus isolates
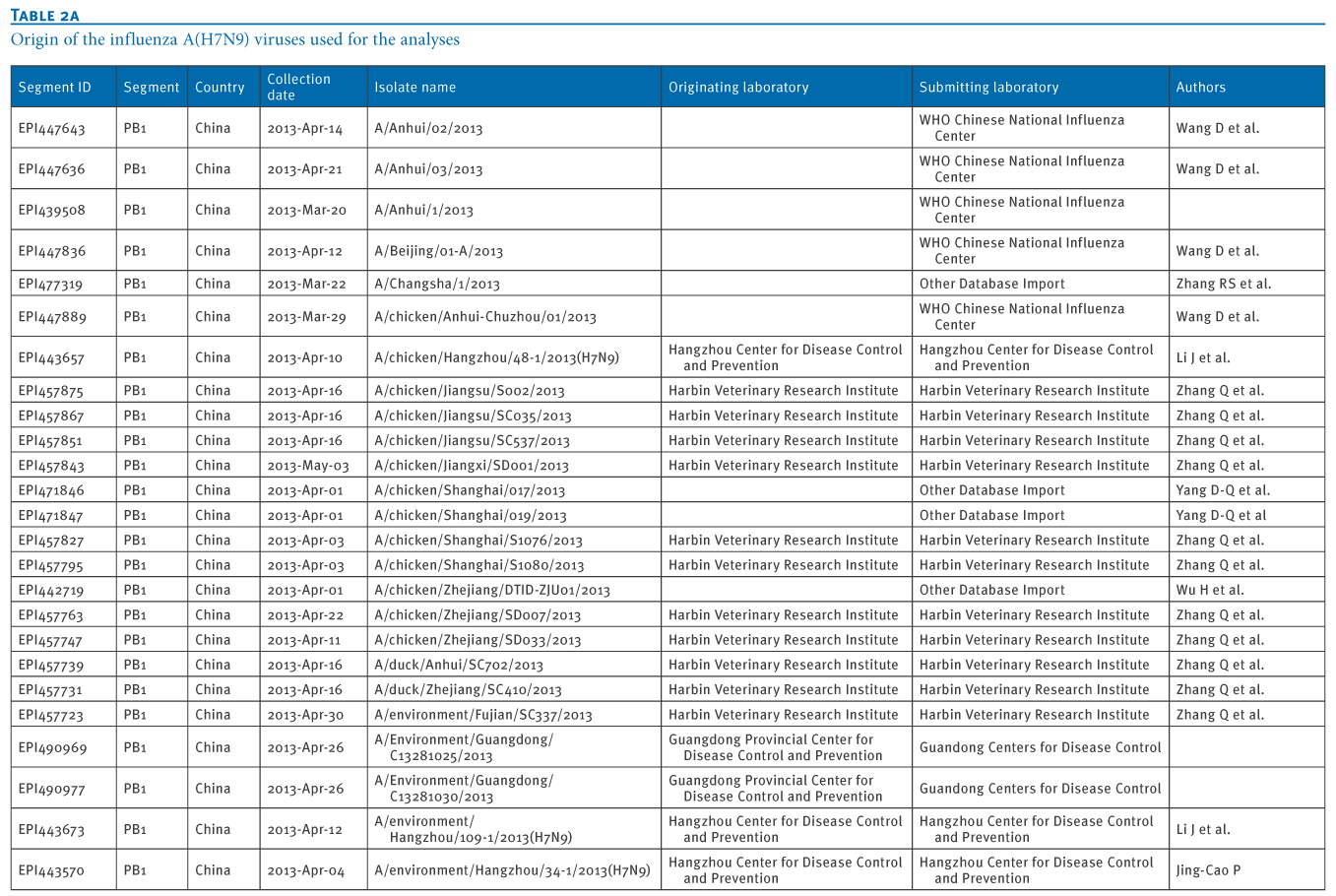
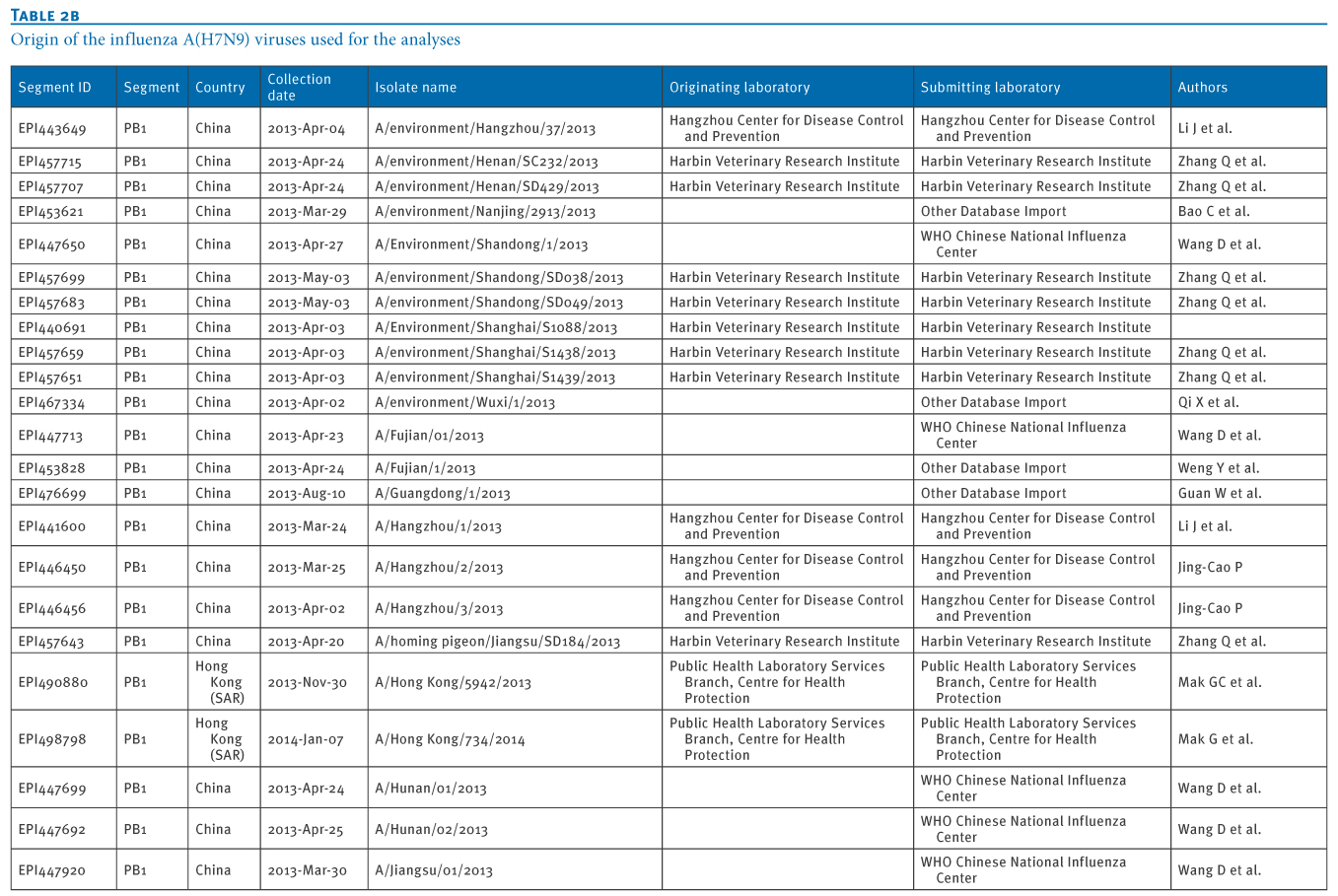
We retrieved 72 PB1 gene sequences of influenza A(H7N9) viruses, isolated from 11 Chinese provinces and cities, from the EpiFlu database of the Global Initiative on Sharing Avian Influenza Data (GISAID) deposited from March 2013 to January 2014 (Tables 1 and 2). In particular, the most recent A(H7N9) virus isolates from Hong Kong were also retrieved, through GISAID (A/Hong Kong/5942/2013 in November 2013 and A/Hong Kong/734/2014 in January 2014).We carried out a Basic Local Alignment Search Tool (BLAST) search to acquire related reference sequences in the National Center for Biotechnology Information (NCBI) Influenza Virus Resource [7]. Multiple alignments of sequences of eight genes of A(H7N9) virus isolates (PB2, PB1, PA, HA, NP, NA, MP, NS) were made using Bio-Edit7.0 software. We then carried out a phylogenetic analysis using MEGA6.1, as previously described [8,9].
______
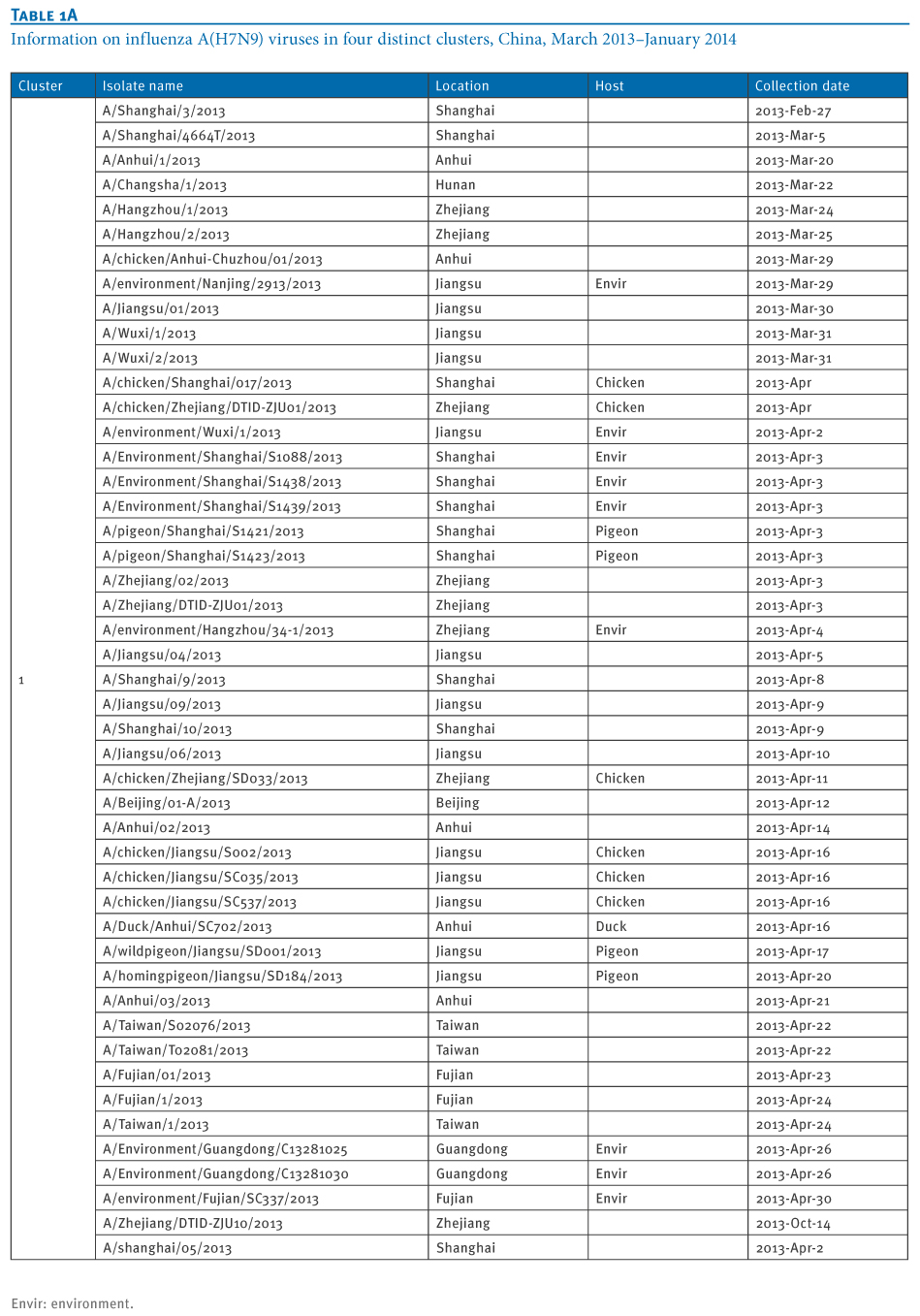
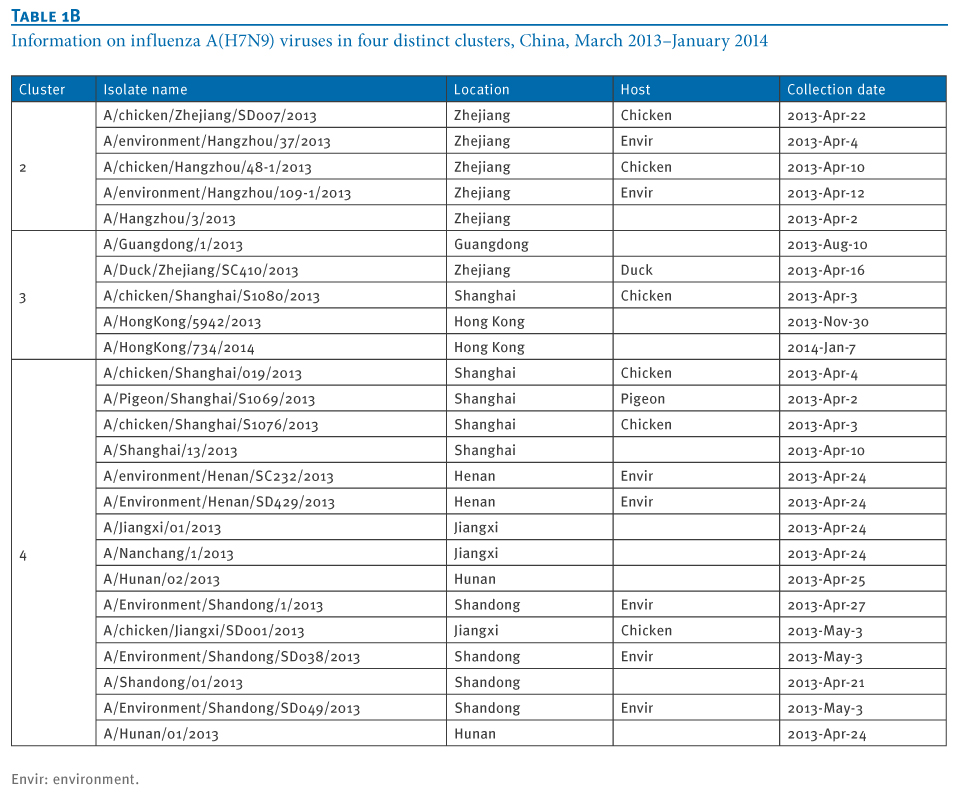
Table 1. Information on influenza A(H7N9) viruses in four distinct clusters, China, March 2013?January 2014


_______
Table 2. Origin of the influenza A(H7N9) viruses used for the analyses



_______
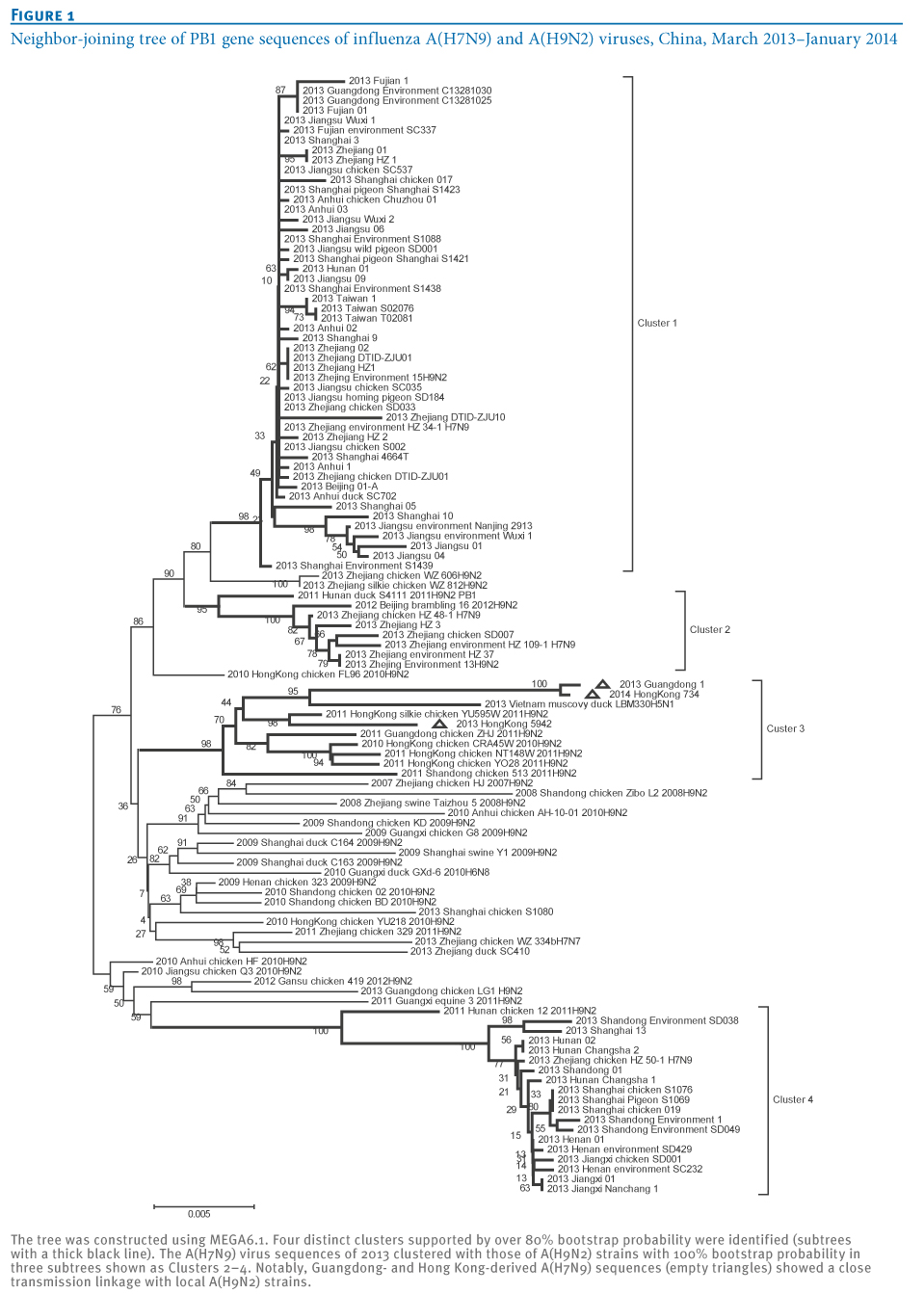
In order to generate a neighbor-joining tree, the statistical robustness of the tree and the reliability of the branching patterns were confirmed by bootstrapping (1,000 replicates) and the effective transmission linkage was supported by a bootstrap value over 80% at the tree node. In accordance with previous studies reporting the virus as a triple reassortant A(H7N9) [2-4], we also observed that all A(H7N9) virus strains analysed, including the latest strains from Hong Kong (Hong Kong strains 5942 and 734), were part of one large cluster in an HA and NA gene-derived neighbor-joining tree (data not shown). However, analysis of six internal genes originating from influenza A(H9N2) virus identified multiple effective A(H7N9) clusters in PB2, PB1, NP, MP gene-derived neighbor-joining trees. As previously described, there is frequent PB2-PB1-PA-NP co-segregation during avian influenza virus reassortment [10]. Clusters of A(H7N9) consistent with this were observed in PB2, PB1 and NP gene-derived neighbor-joining trees (data not shown). Therefore, we then performed further phylogenetic analysis of A(H7N9) and A(H9N2) PB1 gene sequences.
At least four distinct clusters of A(H7N9) virus isolates were identified in a PB1 gene-derived neighbor-joining tree by high bootstrap value (>80%) (Figure 1). Cluster 1 containing poultry- or human-derived A(H7N9) virus isolates represents the earliest infections (shown by the collection date in Tables 1 and 2) and covers the majority of A(H7N9) virus infections in 2013, while the other three clusters indicate close phylogenetic links between A(H7N9) and A(H9N2) strains.
______
Figure 1. Neighbor-joining tree of PB1 gene sequences of influenza A(H7N9) and A(H9N2) viruses, China, March 2013?January 2014

_______
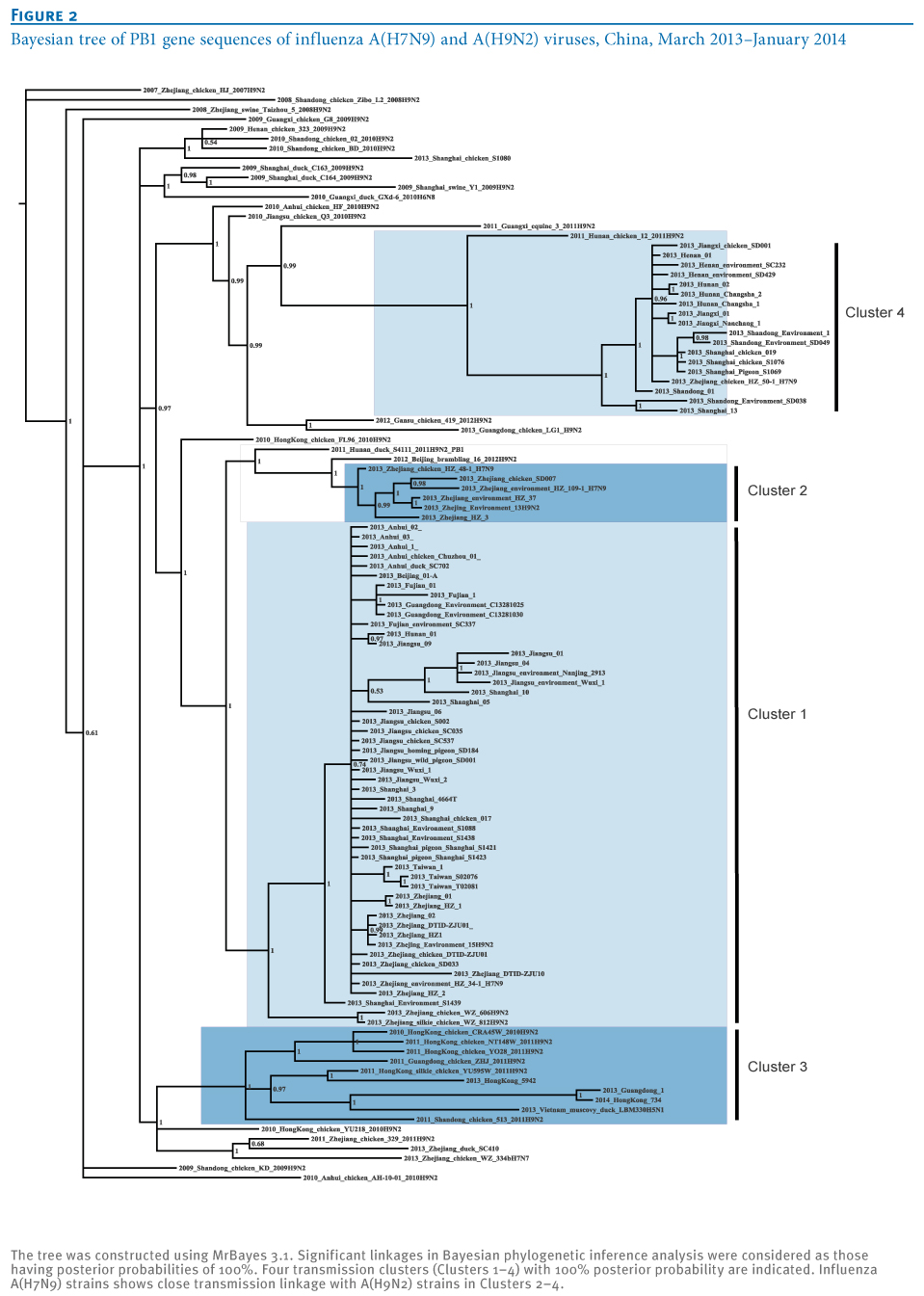
Generally, identification of distinct transmission clusters should meet the following criteria: a phylogenetic clade supported by both high bootstrap values (>80%) in a neighbor-joining tree and a posterior probability value of 1 at the Bayesian tree node [11,12]. For this purpose, a Bayesian phylogenetic inference was subsequently performed to confirm the distinct clusters of A(H7N9) isolates using MrBayes 3.1 as previously described [8,9]. As expected, the same four clusters (100% probability; posterior probability=1) were also seen in the Bayesian tree (Figure 2), as well as in the neighbor-joining tree (Figure 1), which further identified the effective transmission linkages inside these clusters (Figure 2).
______
Figure 2. Bayesian tree of PB1 gene sequences of influenza A(H7N9) and A(H9N2) viruses, China, March 2013?January 2014

_______
Characterisation of transmission clusters
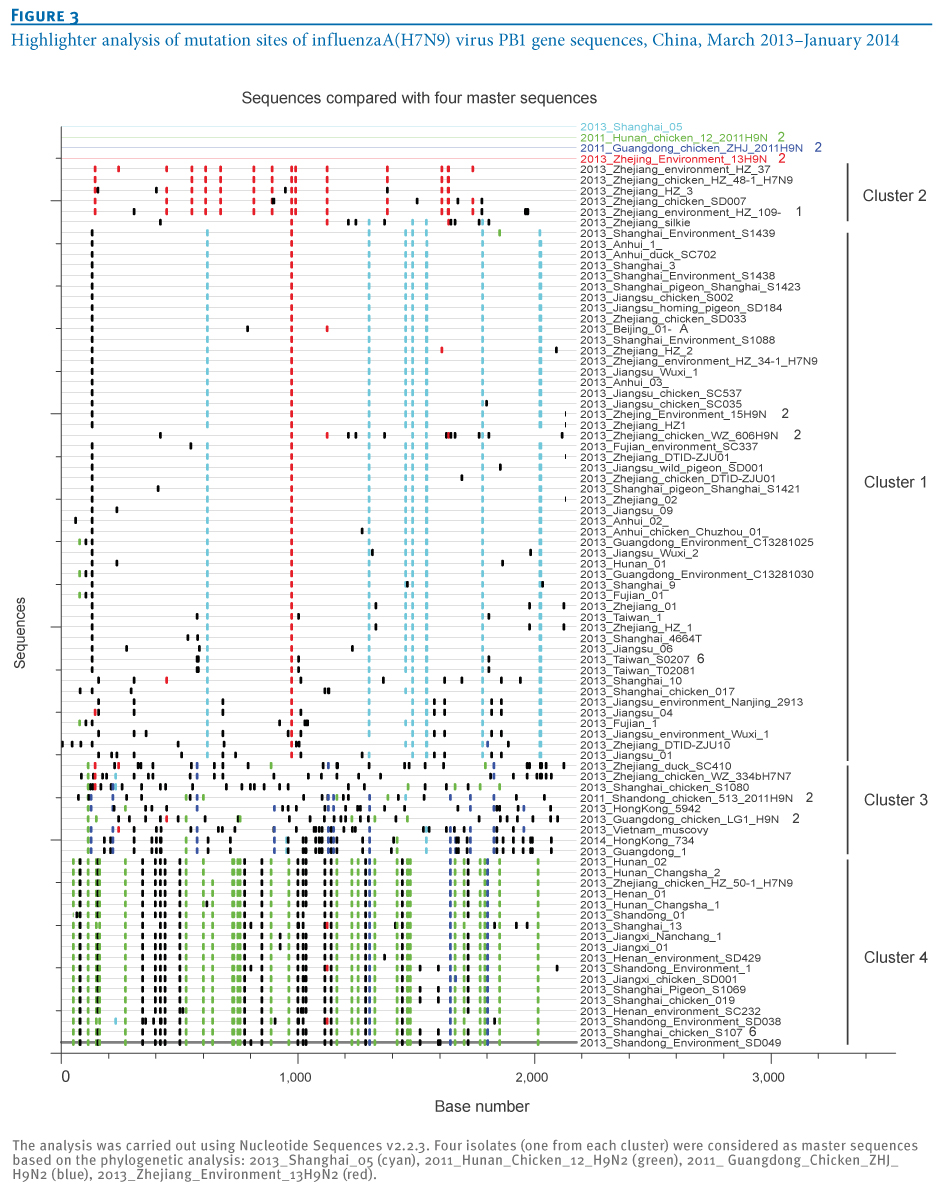
To further characterise the four transmission clusters of influenza A(H7N9) virus isolates, the mutation sites of the viral PB1 gene sequences were highlighted using Nucleotide Sequences v2.2.3 (Figure3), revealing a distinct DNA mutation pattern of the four transmission clusters. Cluster1 shared the most common mutation sites with Shanghai-derived A(H7N9) strains, while all A(H7N9) strains from the other three clusters carried the most common mutation sites of their local A(H9N2) strains. The A(H7N9) strains of Cluster 2 carried the most common mutation sites of a Zhejiang-derived A(H9N2) strain, whereas the A(H7N9) strains in Clusters 3 and 4 had the most common mutation sites of Guangdong/Hong Kong-derived A(H9N2) and Hunan-derived A(H9N2) strains, respectively. These distinct DNA mutation patterns further identified new reassortments between A(H7N9) isolates and local A(H9N2) strains.
______
Figure 3. Highlighter analysis of mutation sites of influenza A(H7N9) virus PB1 gene sequences, China, March 2013?January 2014

______
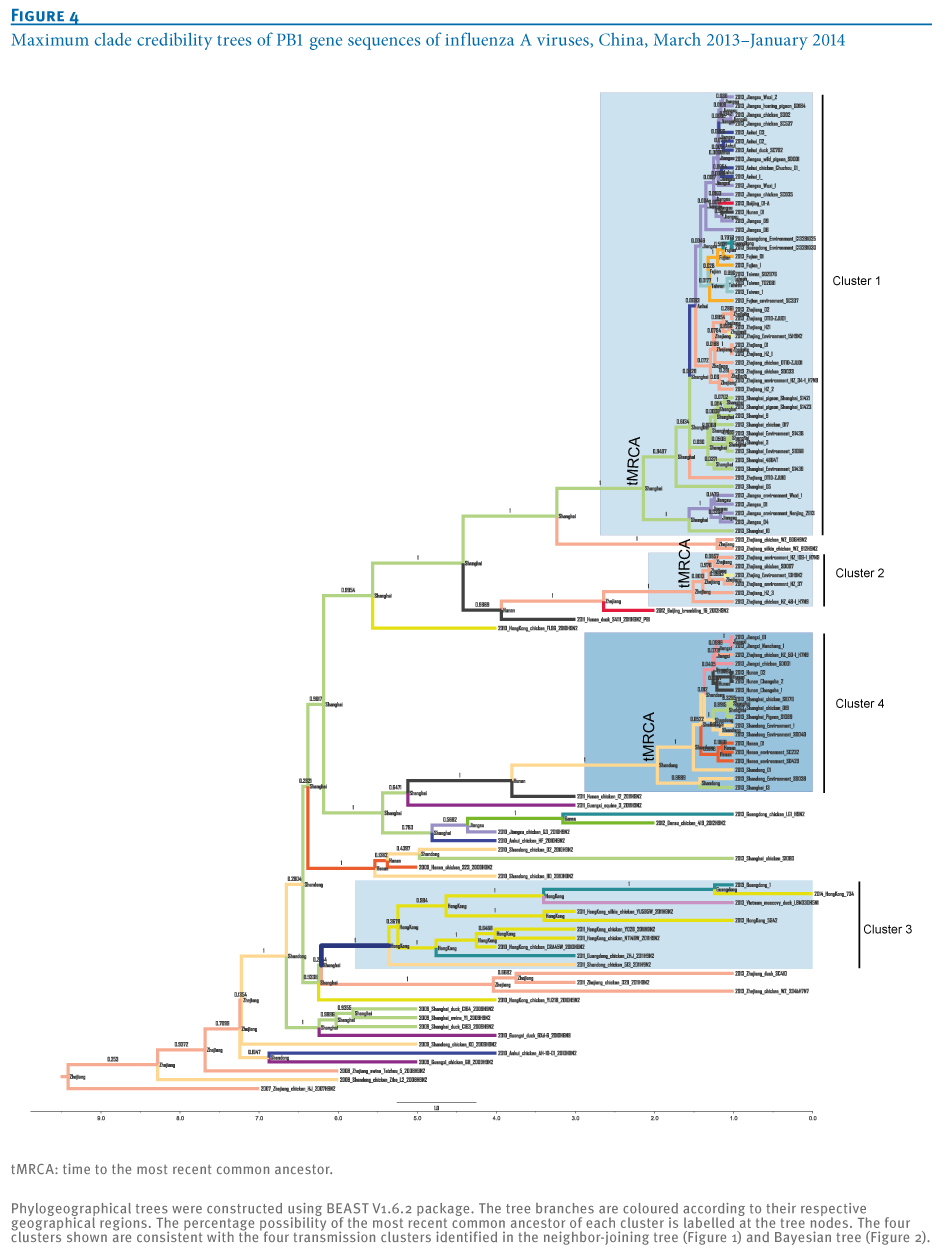
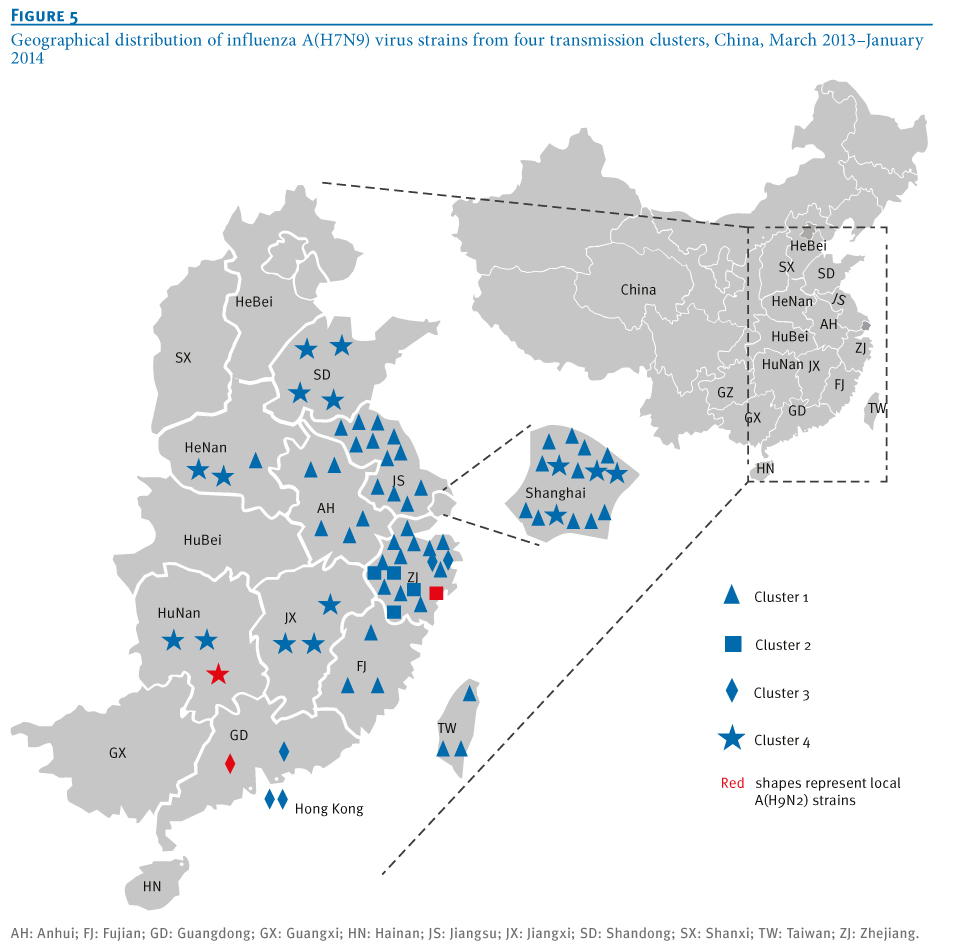
Phylogeographical trees of the influenza A virus PB1 gene sequences were constructed to further confirm the phylogenetic linkage of A(H7N9)and A(H9N2) virus strains using the BEAST V1.6.2 package as described previously [13,14]. The most recent common ancestor of the four clusters was estimated to be from Shanghai-derived strains (Figure 4). In addition, the collection dates of Shanghai A(H7N9) strains were earlier than those of other strains in Clusters 1 and 4 (Tables 1 and 2), suggesting a critical role of Shanghai strains in dissemination of the virus (Figure 5). A new reassortment involving Zhejiang-derived A(H7N9) and Guangdong local A(H9N2) strains formed one independent cluster (Cluster 3), representing the latest and furthest A(H7N9) strains (from the earliest infecting strains in Shanghai) [2] (Figure 4). Moreover, as indicated by Cluster 4 in Figures 4 and 5, the new reassortment of Shanghai A(H7N9)- and Hunan A(H9N2)-derived strains may facilitate further dissemination of A(H7N9) virus from the Yangtze River Delta Economic Zone (including Shanghai, Jiangsu, Zhejiang, Anhui) into neighbouring provinces (such as Shandong, Hunan, Henan and Jiangxi). Meanwhile, Shanghai and Zhejiang A(H7N9) strains were involved in Clusters 3 and 4, indicating that the new reassortment may occur in poultry. Moreover, both the time of the most recent common ancestor and collection date of strains in Clusters 2 and 4 (Figures 4 and 6) showed an obvious delay in comparison with those in Cluster 1, suggesting that reassortment probably occurred during the initial outbreaks. Unlike Clusters 3 and 4, Cluster 2 represents the reassortment between local A(H7N9) and A(H9N2) strains (both from Zhejiang).
______
Figure 4. Maximum clade credibility trees of PB1 gene sequences of influenza A viruses, China, March 2013?January 2014

______
Figure 5. Geographical distribution of influenza A(H7N9) virus strains from four transmission clusters, China, March 2013?January 2014

_______
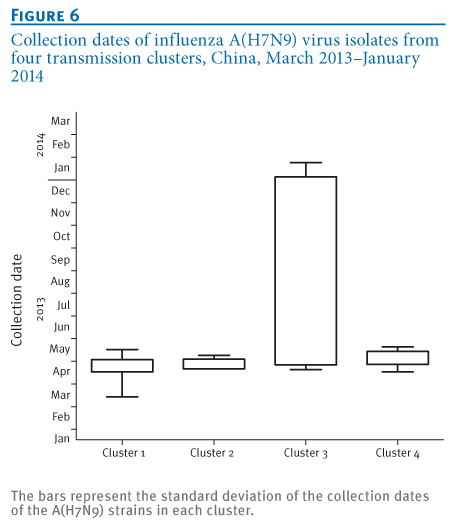
Notably, the distinct time to the most recent common ancestor of Clusters 1, 2 and 4 (Figure 4) is consistent with the time course of collection date of the strains in the three clusters (Figure 6, Tables 1 and 2), suggesting distinct phases for transmission and reassortment of A(H7N9) virus in China. Cluster 1, with the earliest most recent common ancestor (Figure 4), may represent the first wave and main body of the A(H7N9) outbreak during first half of 2013, which facilitated the subsequent reassortment between A(H7N9) and local A(H9N2) strains, as Clusters 2 and 4 indicate.
Additionally, although no time to the most recent common ancestor is indicated for Cluster 3 (Figure 4), all A(H7N9) strains in this cluster have been isolated very recently (Tables 1, and 2, Figure 6), which may represent the latest reassortment of A(H7N9) and A(H9N2) strains. The association between the expanding transmission and appearance of reassortments suggests a tendency for A(H7N9) evolution towards more and more geographical localisation. In addition, Shanghai or Zhejiang poultry-derived A(H7N9) strains may also play active roles in the process of reassortment and localisation (Tables 1 and 2).
_______
Figure 6. Collection dates of influenza A(H7N9) virus isolates from four transmission clusters, China, March 2013?January 2014

______
Discussion
Our analysis revealed dynamic reassortments between influenza A(H7N9) and A(H9N2) viruses since the outbreak of A(H7N9) virus infection in March 2013.To some extent, the continuous transmission of H7N9 in Chinese poultry has led to increasing diversity and new reassortment of A(H7N9) with local A(H9N2) strains. Our findings suggest that the re-emerged H7N9 infections may be triggered by new reassortment strains, such as those in the Guangdong/Hong Kong transmission of Cluster 3. In this regard, these infections may have implications for the traditional strategies of drug and vaccine development targeted against HA and NA genes [15].In particular, the new reassortments generated by A(H7N9) and local A(H9N2) strains may produce avian influenza virus strains that are more adaptive and have a higher pathogenicity in humans [16], emphasising the importance of continuously monitoring the A(H7N9) epidemic.
To date, 127 cases of A(H7N9) virus infections have been reported in January 2014, almost the same number as reported in the spring of 2013 (n=133) [5,6]. Notably, Zhejiang and Guangdong provinces and the Shanghai metropolitan area, where new reassortment of A(H7N9) strains is being identified, have been the worst affected regions in China in 2014 [1,17,18]. Although the case-fatality rate in January 2014 (24%, 31/127) is not higher than that seen in the spring of 2013 (29%, 39/133) [5,6], the rapidly increasing number of cases of A(H7N9) virus infection in these three regions may raise concerns as to whether there is an association between circulation of the new A(H7N9) reassortment strains identified and accelerated transmission of A(H7N9) virus in humans. Therefore, it is of the utmost importance to monitor the risk of a potential pandemic initiated by various influenza virus strains.
______
Acknowledgements
We acknowledge the authors, originating and submitting laboratories of the sequences from GISAID?s EpiFlu Database on which this research is based (see Table 2). All submitters of the data may be contacted directly via the GISAID website www.gisaid.org. This work was supported by Chinese National Grand Program on Key Infectious Disease Control (2012ZX10004-211), Shanghai Municipal Commission of Health and Family Planning (2013QLG003) and 985 program at Fudan University (EZF101606/018-019).
Conflict of interest
None declared.
Authors? contributions
Z.M. and J.X. conceived and designed the experiments. Z.M. performed the experiments and analysed the data. Z.Y., Y.H. and X.Z. contributed reagents/materials/analysis tools. Z.M. and R.H. wrote the paper.
References
-
-------
Eurosurveillance, Volume 19, Issue 6, 13 February 2014
Rapid communications
Possible pandemic threat from new reassortment of influenza A(H7N9) virus in China
Z Meng<SUP>1</SUP>, R Han<SUP>2</SUP>, Y Hu<SUP>1</SUP>, Z Yuan<SUP>1</SUP>, S Jiang<SUP>1</SUP>, X Zhang<SUP>1</SUP><SUP>,3</SUP>, J Xu <SUP>1</SUP><SUP>,3</SUP>
<SUP></SUP>
<SUP>1</SUP>Shanghai Public Health Clinical Center and Institutes of Biomedical Sciences, Key Laboratory of Medical Molecular Virology of Ministry of Education/Health, Shanghai Medical College, Fudan University, Shanghai, China - 2 Department of Cell and Molecular Physiology, Loyola University Chicago Health Sciences Division, Maywood, IL, United States - 3 State Key Laboratory for Infectious Disease Prevention and Control, China Center for Disease Control and Prevention, Beijing, China
_____
Citation style for this article: Meng Z, Han R, Hu Y, Yuan Z, Jiang S, Zhang X, Xu J. Possible pandemic threat from new reassortment of influenza A(H7N9) virus in China. Euro Surveill. 2014;19(6):pii=20699. Available online: http://www.eurosurveillance.org/View...rticleId=20699
Date of submission: 29 January 2014
_____
Avian influenza A(H7N9) virus re-emerged in China in December 2013, after a decrease in the number of new cases during the preceding six months. Reassortment between influenza A(H7N9) and local H9N2 strains has spread from China's south-east coast to other regions. Three new reassortments of A(H7N9) virus were identified by phylogenetic analysis: between A(H7N9) and Zhejiang-derived strains, Guangdong/Hong Kong-derived strains or Hunan-derived A(H9N2) strains. Our findings suggest there is a possible risk that a pandemic could develop.
________
Recent re-emerged influenza A(H7N9) virus infections in China ? especially the rapid outbreak in Zhejiang province in December 2013, involving 60 cases [1] ? have raised concerns. Although several reports described the genetic characteristics of the virus [2-4], little is known about its further evolution after the initial outbreak in March 2013 [2] and the current re-emergence. As of 31 January 2014, there were a total of 260 cases: 127 of these have occurred in 2014 [5,6]. Cases have been reported from Zhejiang, Guangdong and Jiangsu provinces, Shanghai metropolitan area and Hong Kong in 2014 [6].
It is important to know whether new variants or lineages of influenza A(H7N9) virus are responsible for this re-emergence of the virus. In this study, four lineages and three new reassortments of A(H7N9) virus were identified by phylogenetic analysis and DNA mutation analysis of the PB1 gene.
Sequences analysis of PB1 genes from influenza A(H7N9) virus isolates
We retrieved 72 PB1 gene sequences of influenza A(H7N9) viruses, isolated from 11 Chinese provinces and cities, from the EpiFlu database of the Global Initiative on Sharing Avian Influenza Data (GISAID) deposited from March 2013 to January 2014 (Tables 1 and 2). In particular, the most recent A(H7N9) virus isolates from Hong Kong were also retrieved, through GISAID (A/Hong Kong/5942/2013 in November 2013 and A/Hong Kong/734/2014 in January 2014).We carried out a Basic Local Alignment Search Tool (BLAST) search to acquire related reference sequences in the National Center for Biotechnology Information (NCBI) Influenza Virus Resource [7]. Multiple alignments of sequences of eight genes of A(H7N9) virus isolates (PB2, PB1, PA, HA, NP, NA, MP, NS) were made using Bio-Edit7.0 software. We then carried out a phylogenetic analysis using MEGA6.1, as previously described [8,9].
______
Table 1. Information on influenza A(H7N9) viruses in four distinct clusters, China, March 2013?January 2014
_______
Table 2. Origin of the influenza A(H7N9) viruses used for the analyses
_______
In order to generate a neighbor-joining tree, the statistical robustness of the tree and the reliability of the branching patterns were confirmed by bootstrapping (1,000 replicates) and the effective transmission linkage was supported by a bootstrap value over 80% at the tree node. In accordance with previous studies reporting the virus as a triple reassortant A(H7N9) [2-4], we also observed that all A(H7N9) virus strains analysed, including the latest strains from Hong Kong (Hong Kong strains 5942 and 734), were part of one large cluster in an HA and NA gene-derived neighbor-joining tree (data not shown). However, analysis of six internal genes originating from influenza A(H9N2) virus identified multiple effective A(H7N9) clusters in PB2, PB1, NP, MP gene-derived neighbor-joining trees. As previously described, there is frequent PB2-PB1-PA-NP co-segregation during avian influenza virus reassortment [10]. Clusters of A(H7N9) consistent with this were observed in PB2, PB1 and NP gene-derived neighbor-joining trees (data not shown). Therefore, we then performed further phylogenetic analysis of A(H7N9) and A(H9N2) PB1 gene sequences.
At least four distinct clusters of A(H7N9) virus isolates were identified in a PB1 gene-derived neighbor-joining tree by high bootstrap value (>80%) (Figure 1). Cluster 1 containing poultry- or human-derived A(H7N9) virus isolates represents the earliest infections (shown by the collection date in Tables 1 and 2) and covers the majority of A(H7N9) virus infections in 2013, while the other three clusters indicate close phylogenetic links between A(H7N9) and A(H9N2) strains.
______
Figure 1. Neighbor-joining tree of PB1 gene sequences of influenza A(H7N9) and A(H9N2) viruses, China, March 2013?January 2014
_______
Generally, identification of distinct transmission clusters should meet the following criteria: a phylogenetic clade supported by both high bootstrap values (>80%) in a neighbor-joining tree and a posterior probability value of 1 at the Bayesian tree node [11,12]. For this purpose, a Bayesian phylogenetic inference was subsequently performed to confirm the distinct clusters of A(H7N9) isolates using MrBayes 3.1 as previously described [8,9]. As expected, the same four clusters (100% probability; posterior probability=1) were also seen in the Bayesian tree (Figure 2), as well as in the neighbor-joining tree (Figure 1), which further identified the effective transmission linkages inside these clusters (Figure 2).
______
Figure 2. Bayesian tree of PB1 gene sequences of influenza A(H7N9) and A(H9N2) viruses, China, March 2013?January 2014
_______
Characterisation of transmission clusters
To further characterise the four transmission clusters of influenza A(H7N9) virus isolates, the mutation sites of the viral PB1 gene sequences were highlighted using Nucleotide Sequences v2.2.3 (Figure3), revealing a distinct DNA mutation pattern of the four transmission clusters. Cluster1 shared the most common mutation sites with Shanghai-derived A(H7N9) strains, while all A(H7N9) strains from the other three clusters carried the most common mutation sites of their local A(H9N2) strains. The A(H7N9) strains of Cluster 2 carried the most common mutation sites of a Zhejiang-derived A(H9N2) strain, whereas the A(H7N9) strains in Clusters 3 and 4 had the most common mutation sites of Guangdong/Hong Kong-derived A(H9N2) and Hunan-derived A(H9N2) strains, respectively. These distinct DNA mutation patterns further identified new reassortments between A(H7N9) isolates and local A(H9N2) strains.
______
Figure 3. Highlighter analysis of mutation sites of influenza A(H7N9) virus PB1 gene sequences, China, March 2013?January 2014
______
Phylogeographical trees of the influenza A virus PB1 gene sequences were constructed to further confirm the phylogenetic linkage of A(H7N9)and A(H9N2) virus strains using the BEAST V1.6.2 package as described previously [13,14]. The most recent common ancestor of the four clusters was estimated to be from Shanghai-derived strains (Figure 4). In addition, the collection dates of Shanghai A(H7N9) strains were earlier than those of other strains in Clusters 1 and 4 (Tables 1 and 2), suggesting a critical role of Shanghai strains in dissemination of the virus (Figure 5). A new reassortment involving Zhejiang-derived A(H7N9) and Guangdong local A(H9N2) strains formed one independent cluster (Cluster 3), representing the latest and furthest A(H7N9) strains (from the earliest infecting strains in Shanghai) [2] (Figure 4). Moreover, as indicated by Cluster 4 in Figures 4 and 5, the new reassortment of Shanghai A(H7N9)- and Hunan A(H9N2)-derived strains may facilitate further dissemination of A(H7N9) virus from the Yangtze River Delta Economic Zone (including Shanghai, Jiangsu, Zhejiang, Anhui) into neighbouring provinces (such as Shandong, Hunan, Henan and Jiangxi). Meanwhile, Shanghai and Zhejiang A(H7N9) strains were involved in Clusters 3 and 4, indicating that the new reassortment may occur in poultry. Moreover, both the time of the most recent common ancestor and collection date of strains in Clusters 2 and 4 (Figures 4 and 6) showed an obvious delay in comparison with those in Cluster 1, suggesting that reassortment probably occurred during the initial outbreaks. Unlike Clusters 3 and 4, Cluster 2 represents the reassortment between local A(H7N9) and A(H9N2) strains (both from Zhejiang).
______
Figure 4. Maximum clade credibility trees of PB1 gene sequences of influenza A viruses, China, March 2013?January 2014
______
Figure 5. Geographical distribution of influenza A(H7N9) virus strains from four transmission clusters, China, March 2013?January 2014
_______
Notably, the distinct time to the most recent common ancestor of Clusters 1, 2 and 4 (Figure 4) is consistent with the time course of collection date of the strains in the three clusters (Figure 6, Tables 1 and 2), suggesting distinct phases for transmission and reassortment of A(H7N9) virus in China. Cluster 1, with the earliest most recent common ancestor (Figure 4), may represent the first wave and main body of the A(H7N9) outbreak during first half of 2013, which facilitated the subsequent reassortment between A(H7N9) and local A(H9N2) strains, as Clusters 2 and 4 indicate.
Additionally, although no time to the most recent common ancestor is indicated for Cluster 3 (Figure 4), all A(H7N9) strains in this cluster have been isolated very recently (Tables 1, and 2, Figure 6), which may represent the latest reassortment of A(H7N9) and A(H9N2) strains. The association between the expanding transmission and appearance of reassortments suggests a tendency for A(H7N9) evolution towards more and more geographical localisation. In addition, Shanghai or Zhejiang poultry-derived A(H7N9) strains may also play active roles in the process of reassortment and localisation (Tables 1 and 2).
_______
Figure 6. Collection dates of influenza A(H7N9) virus isolates from four transmission clusters, China, March 2013?January 2014
______
Discussion
Our analysis revealed dynamic reassortments between influenza A(H7N9) and A(H9N2) viruses since the outbreak of A(H7N9) virus infection in March 2013.To some extent, the continuous transmission of H7N9 in Chinese poultry has led to increasing diversity and new reassortment of A(H7N9) with local A(H9N2) strains. Our findings suggest that the re-emerged H7N9 infections may be triggered by new reassortment strains, such as those in the Guangdong/Hong Kong transmission of Cluster 3. In this regard, these infections may have implications for the traditional strategies of drug and vaccine development targeted against HA and NA genes [15].In particular, the new reassortments generated by A(H7N9) and local A(H9N2) strains may produce avian influenza virus strains that are more adaptive and have a higher pathogenicity in humans [16], emphasising the importance of continuously monitoring the A(H7N9) epidemic.
To date, 127 cases of A(H7N9) virus infections have been reported in January 2014, almost the same number as reported in the spring of 2013 (n=133) [5,6]. Notably, Zhejiang and Guangdong provinces and the Shanghai metropolitan area, where new reassortment of A(H7N9) strains is being identified, have been the worst affected regions in China in 2014 [1,17,18]. Although the case-fatality rate in January 2014 (24%, 31/127) is not higher than that seen in the spring of 2013 (29%, 39/133) [5,6], the rapidly increasing number of cases of A(H7N9) virus infection in these three regions may raise concerns as to whether there is an association between circulation of the new A(H7N9) reassortment strains identified and accelerated transmission of A(H7N9) virus in humans. Therefore, it is of the utmost importance to monitor the risk of a potential pandemic initiated by various influenza virus strains.
______
Acknowledgements
We acknowledge the authors, originating and submitting laboratories of the sequences from GISAID?s EpiFlu Database on which this research is based (see Table 2). All submitters of the data may be contacted directly via the GISAID website www.gisaid.org. This work was supported by Chinese National Grand Program on Key Infectious Disease Control (2012ZX10004-211), Shanghai Municipal Commission of Health and Family Planning (2013QLG003) and 985 program at Fudan University (EZF101606/018-019).
Conflict of interest
None declared.
Authors? contributions
Z.M. and J.X. conceived and designed the experiments. Z.M. performed the experiments and analysed the data. Z.Y., Y.H. and X.Z. contributed reagents/materials/analysis tools. Z.M. and R.H. wrote the paper.
References
- Health and Family Planning Commission of Zhejiang Province. [Daily report for avian flu cases in Zhejiang province]. Hangzhou: Health and Family Planning Commission of Zhejiang Province. Updated 31 Jan 2014. [Accessed 9 Feb 2014]. Chinese. Available from: http://www.zjwst.gov.cn/col/col371/index.html###
- Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368(20):1888-97. http://dx.doi.org/10.1056/NEJMoa1304459
- Liu D, Shi W, Shi Y, Wang D, Xiao H, Li W, et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses. Lancet. 2013;381(9881):1926-32.
http://dx.doi.org/10.1016/S0140-6736(13)60938-1 - Hu Y, Lu S, Song Z, Wang W, Hao P, Li J, et al. Association between adverse clinical outcome in human disease caused by novel influenza A H7N9 virus and sustained viral shedding and emergence of antiviral resistance. Lancet. 2013;381(9885):2273-9.
http://dx.doi.org/10.1016/S0140-6736(13)61125-3 - National Healthy and Family Planning Commission of the People's Republic of China. [The preliminary achievements in prevention and therapy of H7N9 human infections]. Beijing: National Healthy and Family Planning Commission of the People's Republic of China. Updated 9 Jun 2013. [Accessed 9 Feb 2014]. Chinese. Available from: http://www.moh.gov.cn/zhuzhan/yqxx%20/201306/8a945b75a8b742f585b942ee4e4f3155.shtml
- National Healthy and Family Planning Commission of the People's Republic of China. [January 2014 national overview of legal infectious diseases]. Beijing: National Healthy and Family Planning Commission of the People's Republic of China. Updated 31 Jan 2014. [Accessed 9 Feb 2014]. Chinese. Available from: http://www.nhfpc.gov.cn/jkj/s3578/201402/9c8e60933cd046e090327184b26f8349.shtml
- Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, et al.The influenza virus resource at the National Center for Biotechnology Information. J Virol. 2008; 82(2):596-601. http://dx.doi.org/10.1128/JVI.02005-07
- Posada D, Buckley TR. Model selection and model averaging in phylogenetics: advantages of Akaike Information Criterion and Bayesian approaches over likelihood ratio tests. Syst Biol. 2004;53(5):793-808. http://dx.doi.org/10.1080/10635150490522304
- Yang Z, Rannala B. Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo method. Mol Biol Evol. 1997;14(7):717-24. http://dx.doi.org/10.1093/oxfordjournals.molbev.a025811
- Chan JM,Carlsson G,Rabadan R. Topology of viral evolution.Proc Natl Acad Sci U S A. 2013;110(46):18566-71. http://dx.doi.org/10.1073/pnas.1313480110
- Chalmet K, Staelens D, Blot S, Dinakis S, Pelgrom J, Plum J, et al. Epidemiological study of phylogenetic transmission clusters in a local HIV-1 epidemic reveals distinct differences between subtype B and non-B infections. BMC Infect Dis. 2010;10:262.
http://dx.doi.org/10.1186/1471-2334-10-262 - Zehender G, Ebranati E, Lai A, Santoro MM, Alteri C, Giuliani M, et al. Population dynamics of HIV-1 subtype B in a cohort of men-having-sex-with-men in Rome, Italy. J Acquir Immune Defic Syndr. 2010;55(2):156-60. http://dx.doi.org/10.1097/QAI.0b013e3181eb3002
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
http://dx.doi.org/10.1186/1471-2148-7-214 - Lemey P, Suchard M, Rambaut A. Reconstructing the initial global spread of a human influenza pandemic: a Bayesian spatial-temporal model for the global spread of H1N1pdm.PLoS Curr. 2009;1:RRN1031. http://dx.doi.org/10.1371/currents.RRN1031
- Mei L, Song P, Tang Q, Shan K, Tobe RG, SelotlegengL,et al. Changes in and shortcomings of control strategies, drug stockpiles, and vaccine development during outbreaks of avian influenza A H5N1, H1N1, and H7N9 among humans.Biosci Trends. 2013;7(2):64-76.
- Liu Q, Lu L, Sun Z, Chen GW, Wen Y, Jiang S. Genomic signature and protein sequence analysis of a novel influenza A (H7N9) virus that causes an outbreak in humans in China. Microbes Infect. 2013;15(6-7):432-9. http://dx.doi.org/10.1016/j.micinf.2013.04.004
- Health Department of Guangdong Province.[Daily report of new H7N9 cases in Guangdong province: 2 new confirmed cases of human infection with H7N9 avian influenza]. Guangzhou: Health Department of Guangdong Province. Updated 9Feb2014.[Accessed 9 Feb 2014]. Chinese. Available from: http://www.gdwst.gov.cn/a/zwxw/2014020911312.html
- Shanghai Municipal Commission of Healthy and Family Planning.[Daily report of new H7N9 cases in Shanghai: 1 new confirmed case of human infection with H7N9 avian influenza]. Shanghai: Shanghai Municipal Commission of Healthy and Family Planning. Updated 23 Jan 2014. Press release. [Accessed 9 Feb 2014]. Chinese. Available from: http://www.wsjsw.gov.cn/wsj/n422/n424/u1ai132467.html
-
-------
Comment