 Tweet
Tweet
Hat tip Mary Wilson and Sharon Sanders
Preliminary report on genomic epidemiology of the 2024 H5N1 influenza A virus outbreak in U.S. cattle (Part 1 of 2) - Virological

Excerpts from this report:
...
Findings
Below, we list some noteworthy preliminary findings, based on analyses of the above sequence data and metadata.
1. A reassortment event within North American avian H5N1 2.3.4.4b viruses occurred shortly before the start of the cattle outbreak.
The cattle sequences are all Genotype B3.13 [see GitHub - USDA-VS/GenoFLU: Influenza data pipeline to automate genotyping assignment 4 for an explanation of genotypes].
This genotype is a reassortant between the Eurasian panzootic H5N1 genotype and low pathogenicity North American genotypes first seen in late 2023.
...
Genotype B3.13 differs from the virus seen in a recent outbreak where H5N1 2.3.4.4b influenza A virus spilled over from poultry to goats. The outbreak in goats was unrelated to the current cattle outbreak.
...
2. The cattle outbreak likely had a single origin from the avian H5N1 reservoir.
To determine whether the cattle outbreak arose from a single origin, we separately inferred maximum likelihood trees for each genome segment. We find that the viruses sampled from cattle form a monophyletic clade in each segment (Figures 2 & 3), consistent with a single introduction of H5N1 into cows and indicative of cattle-to-cattle spread.
...
3. The H5N1 outbreak in cattle likely went undetected and unidentified for an extended period and is now several months old.
...
We estimated the median time to the most recent common ancestor (tMRCA) of the cattle clade as 18 January 2024 (95% HPD: [11 Dec 2023, 18 Feb 2023]). We estimated the tMRCA of the cattle clade and a related human virus as 22 November 2023 (95% HPD: [26 September 2023, 19 January 2024]). And we estimated the median tMRCA of the cattle clade and avian H5N1 as 13 November 2023 (95% HPD: [25 Sep 2023, 3 Jan 2024]).
Hence, the jump from the avian reservoir into cattle likely happened between ~13 November 2023 and ~18 January 2024, meaning that the virus may have been circulating in cattle for up to 5 months before H5N1 was identified in them (Figures 5 & 6).
...
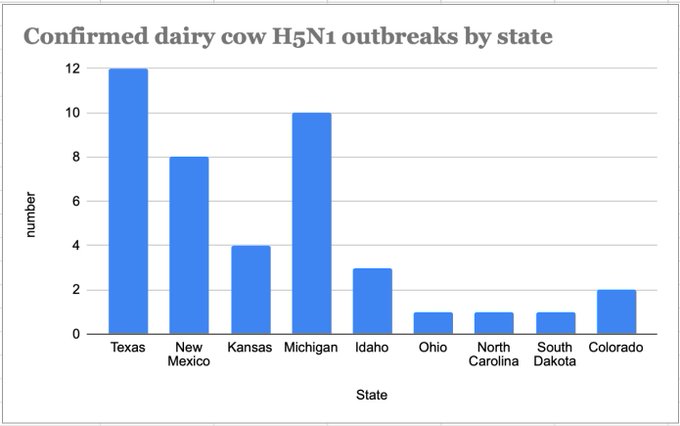
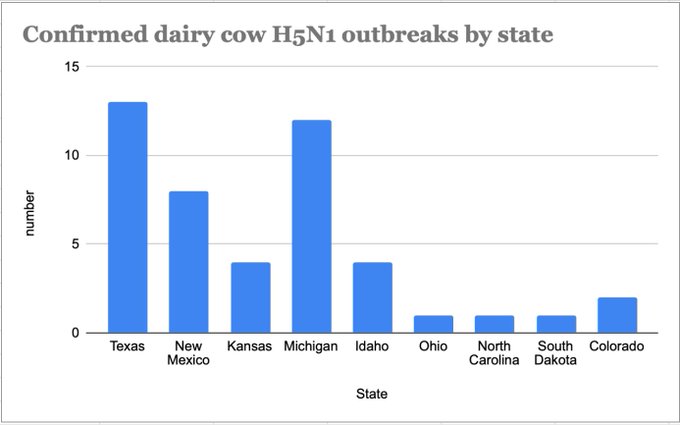
4. The cattle outbreak may have originated in Texas.
The phylogenetic tree is consistent with an origin of the outbreak in Texas, where the first ill and first infected cattle were reported: the basal diversity on the tree is sampled in Texas (Figures 4 & 6).
...
It cannot be ruled out that genotype B3.13 is especially prone to jumping into cattle and other mammals. Both the cattle H5N1 clade and a closely related virus sampled from an individual reportedly exposed on a dairy farm (see section 6, below) are from the same genotype and may have had separate origins from the primarily avian H5N1 2.3.4.4b reservoir.
...
6. The original cattle H5N1 virus’s HA was not adapted to a human-like receptor.
In the hemagglutinin (HA), the protein that must bind to the host’s cell-surface sialic acid residues for the virus to gain entry to the host cell, the closest-related sequences to the cattle H5N1 HAs come from wild birds (a Canada goose and a peregrine falcon.) There are no amino acid differences between the HAs of these wild bird sequences and the earliest sequences from cattle.
This suggests that the first cattle sequences possessed no (pre)adaptation to mammalian cell-surface receptors. Non-synonymous changes in HA have been acquired by small clades of sequences from cattle and cats nested within the cattle clade (n<=11); however, the significance of these mutations is not well understood. Although mutations have been seen in the polymerase of viruses from cattle, similar mutations have been seen in avian viruses infecting humans and other mammals without leading to sustained transmission. Without changes in HA affecting receptor binding, the risk of the virus becoming transmissible between humans is low. The lack of HA changes may also suggest (at least with the current tissue tropism) that there is not strong selective pressure to change receptor binding, suggesting ‘avian-like’ α-2,3-linked sialic acids are abundant in the main sites of replication in these animals. However, this is also true for dogs and pigs, which in the short term do not strongly select for such changes, yet in the longer term, avian-origin H1N1 and H3N2 viruses in these species gradually adapt to ‘human-like’ α-2,6-linked sialic acids(37) 3(38) 2
7. H5N1 is transmitting from cattle back into wild birds, poultry, cats, and other species.
Several sequences from wild birds (e.g., blackbird, grackle), poultry, domestic cats, and other wildlife (e.g., raccoon) are nested within the main cattle clade of sequences across each genome segment (Figures 2 & 3; see Figure 7 for concatenated genome tree). This tree topology suggests that the virus may be spilling back from cows into other host species, which is consistent with prior reports of virus transmission between cattle and poultry at individual farms(39, 40) 1 [https://www.doi.org/10.1126/science.zoo2sbi 5]and from cows into cats (15) 2 .
While most “spillbacks” from cattle were observed in Texas, one cattle-to-poultry transmission was observed in Michigan (clustering with Michigan cattle viruses), and another spillback to raccoons occurred in New Mexico (clustering with New Mexico cattle viruses).
Whether any onward cat-to-cat or poultry-to-poultry transmission occurred following these spillbacks is difficult to resolve at this time. But these data suggest that H5N1 transmission in cattle is extensive enough to initiate outbreaks in other host species.
...
There have been at least two independent spillbacks to domestic (presumably barn) cats in Texas, most likely due to the consumption of raw milk from infected dairy cattle.
...
8. A virus closely related to, but distinct from, those sampled from cattle was sampled from an individual who was reportedly a dairy farm worker.
...
Therefore, we can’t currently resolve whether (a) the human sequence is a descendent of an unsampled cattle lineage that branched off early from the (single-origin) cattle H5N1 clade shortly after its establishment, or (b) emerged after a separate jump from the avian reservoir into cattle, and then from cattle to that individual on the farm where the person worked, or (c) is a direct spillover from an avian source. Scenario (c) seems highly unlikely....
There are currently no cattle sequences from the farm where this case occurred (our understanding is that none have been collected there, or that the specific farm might not be known). Nor do we have data on cattle movements which could help distinguish between these possibilities.
...
Preliminary report on genomic epidemiology of the 2024 H5N1 influenza A virus outbreak in U.S. cattle (Part 1 of 2) - Virological
Excerpts from this report:
...
Findings
Below, we list some noteworthy preliminary findings, based on analyses of the above sequence data and metadata.
1. A reassortment event within North American avian H5N1 2.3.4.4b viruses occurred shortly before the start of the cattle outbreak.
The cattle sequences are all Genotype B3.13 [see GitHub - USDA-VS/GenoFLU: Influenza data pipeline to automate genotyping assignment 4 for an explanation of genotypes].
This genotype is a reassortant between the Eurasian panzootic H5N1 genotype and low pathogenicity North American genotypes first seen in late 2023.
...
Genotype B3.13 differs from the virus seen in a recent outbreak where H5N1 2.3.4.4b influenza A virus spilled over from poultry to goats. The outbreak in goats was unrelated to the current cattle outbreak.
...
2. The cattle outbreak likely had a single origin from the avian H5N1 reservoir.
To determine whether the cattle outbreak arose from a single origin, we separately inferred maximum likelihood trees for each genome segment. We find that the viruses sampled from cattle form a monophyletic clade in each segment (Figures 2 & 3), consistent with a single introduction of H5N1 into cows and indicative of cattle-to-cattle spread.
...
3. The H5N1 outbreak in cattle likely went undetected and unidentified for an extended period and is now several months old.
...
We estimated the median time to the most recent common ancestor (tMRCA) of the cattle clade as 18 January 2024 (95% HPD: [11 Dec 2023, 18 Feb 2023]). We estimated the tMRCA of the cattle clade and a related human virus as 22 November 2023 (95% HPD: [26 September 2023, 19 January 2024]). And we estimated the median tMRCA of the cattle clade and avian H5N1 as 13 November 2023 (95% HPD: [25 Sep 2023, 3 Jan 2024]).
Hence, the jump from the avian reservoir into cattle likely happened between ~13 November 2023 and ~18 January 2024, meaning that the virus may have been circulating in cattle for up to 5 months before H5N1 was identified in them (Figures 5 & 6).
...
4. The cattle outbreak may have originated in Texas.
The phylogenetic tree is consistent with an origin of the outbreak in Texas, where the first ill and first infected cattle were reported: the basal diversity on the tree is sampled in Texas (Figures 4 & 6).
...
It cannot be ruled out that genotype B3.13 is especially prone to jumping into cattle and other mammals. Both the cattle H5N1 clade and a closely related virus sampled from an individual reportedly exposed on a dairy farm (see section 6, below) are from the same genotype and may have had separate origins from the primarily avian H5N1 2.3.4.4b reservoir.
...
6. The original cattle H5N1 virus’s HA was not adapted to a human-like receptor.
In the hemagglutinin (HA), the protein that must bind to the host’s cell-surface sialic acid residues for the virus to gain entry to the host cell, the closest-related sequences to the cattle H5N1 HAs come from wild birds (a Canada goose and a peregrine falcon.) There are no amino acid differences between the HAs of these wild bird sequences and the earliest sequences from cattle.
This suggests that the first cattle sequences possessed no (pre)adaptation to mammalian cell-surface receptors. Non-synonymous changes in HA have been acquired by small clades of sequences from cattle and cats nested within the cattle clade (n<=11); however, the significance of these mutations is not well understood. Although mutations have been seen in the polymerase of viruses from cattle, similar mutations have been seen in avian viruses infecting humans and other mammals without leading to sustained transmission. Without changes in HA affecting receptor binding, the risk of the virus becoming transmissible between humans is low. The lack of HA changes may also suggest (at least with the current tissue tropism) that there is not strong selective pressure to change receptor binding, suggesting ‘avian-like’ α-2,3-linked sialic acids are abundant in the main sites of replication in these animals. However, this is also true for dogs and pigs, which in the short term do not strongly select for such changes, yet in the longer term, avian-origin H1N1 and H3N2 viruses in these species gradually adapt to ‘human-like’ α-2,6-linked sialic acids(37) 3(38) 2
7. H5N1 is transmitting from cattle back into wild birds, poultry, cats, and other species.
Several sequences from wild birds (e.g., blackbird, grackle), poultry, domestic cats, and other wildlife (e.g., raccoon) are nested within the main cattle clade of sequences across each genome segment (Figures 2 & 3; see Figure 7 for concatenated genome tree). This tree topology suggests that the virus may be spilling back from cows into other host species, which is consistent with prior reports of virus transmission between cattle and poultry at individual farms(39, 40) 1 [https://www.doi.org/10.1126/science.zoo2sbi 5]and from cows into cats (15) 2 .
While most “spillbacks” from cattle were observed in Texas, one cattle-to-poultry transmission was observed in Michigan (clustering with Michigan cattle viruses), and another spillback to raccoons occurred in New Mexico (clustering with New Mexico cattle viruses).
Whether any onward cat-to-cat or poultry-to-poultry transmission occurred following these spillbacks is difficult to resolve at this time. But these data suggest that H5N1 transmission in cattle is extensive enough to initiate outbreaks in other host species.
...
There have been at least two independent spillbacks to domestic (presumably barn) cats in Texas, most likely due to the consumption of raw milk from infected dairy cattle.
...
8. A virus closely related to, but distinct from, those sampled from cattle was sampled from an individual who was reportedly a dairy farm worker.
...
Therefore, we can’t currently resolve whether (a) the human sequence is a descendent of an unsampled cattle lineage that branched off early from the (single-origin) cattle H5N1 clade shortly after its establishment, or (b) emerged after a separate jump from the avian reservoir into cattle, and then from cattle to that individual on the farm where the person worked, or (c) is a direct spillover from an avian source. Scenario (c) seems highly unlikely....
There are currently no cattle sequences from the farm where this case occurred (our understanding is that none have been collected there, or that the specific farm might not be known). Nor do we have data on cattle movements which could help distinguish between these possibilities.
...













Comment