I disagree with JJackson's comment above. I think it is possible that what we know as COVID-19 is the result of the manipulation of genetic material. I personally do not know but I am keeping an open mind. Time will tell. We should let the science develop. Read through this thread to see some of the published work from both the Wuhan BL 4 lab and the China CDC lab in Wuhan.
One thing is for sure - through science we will know the origins of this pandemic disease. We owe the thousands who have died to find out.
FluTrackers is committed to a search for the truth - wherever it leads.
-
j'ai une question :
pour r?cup?rer des virus dans un lieu, il faut faire des pr?l?vements ou laisser se promener des lign?es animales con?ues pour cela, du type de celles d?crites dans ces articles:

La sous question devient : quid de la r?glementation quant ? la d?tention et des usages de ce type de lign?e ?
En management, on utilise parfois des animaux sentinelles, par exemple pour les influenza,on met des poules avec des canards. Les meilleurs animaux sentinelles, pour les coronavirus, ce ne sont pas alors ceux con?us en laboratoire, pour cela et mis ou il faut ?
Je dis aussi cela car visiblement pour aller chercher des virus dans une grotte, vu les publications, cela a ?t? peut efficient et tr?s long. Faitre vivre des animaux sentinelles dans ces lieux, ce n'est pas plus simple ?Leave a comment:
-
Is considering a genetic-manipulation origin for SARS-CoV-2 a conspiracy theory that must be censored?
Rossana Segreto1#(Ph.D.)
Abstract
The origin of SARS-CoV-2 is still controversial. Comparative genomic analyses have shown that SARS-CoV-2 is likely to be chimeric, most of its sequence being very close to the CoV detected from a bat, whereas its receptor binding domain is almost identical to that of CoV obtained from pangolins. The furin cleavage site in the spike protein of SARS-CoV-2 was previously not identified in other SARS-like CoVs and might have conferred ability to cross species and tissue barriers. Chimeric viruses can be the product of natural recombination or genetic manipulations. The latter could have aimed to identify pangolins as possible intermediate hosts for bat-CoV potentially pathogenic for humans. Theories that consider a possible artificial origin for SARS-CoV-2 are censored as they seem to support conspiracy theories. Researchers have the responsibility to carry out a thorough analysis, beyond any personal research interests, of all possible causes for SARS-CoV-2 emergence for preventing this from happening in the future.
(PDF) Is considering a genetic-manipulation origin for SARS-CoV-2 a conspiracy theory that must be censored?. Available from: https://www.researchgate.net/publica...st_be_censored [accessed Apr 26 2020].Leave a comment:
-
Originally posted by Pathfinder View Post
HIV gp120 and gag discussed above and in retracted study.Clin Dev Immunol. 2006 Jun-Dec;13(2-4):353-60.
Towards a coronavirus-based HIV multigene vaccine.
Eriksson KK1, Makia D, Maier R, Ludewig B, Thiel V.
Author information
Abstract
Human immunodeficiency virus (HIV) infection represents one of the major health threats in the developing world. The costly treatment of infected individuals with multiple highly efficient anti-HIV drugs is only affordable in industrialized countries. Thus, an efficient vaccination strategy is required to prevent the further spread of the infection. The molecular biology of coronaviruses and particular features of the human coronavirus 229E (HCoV 229E) indicate that HCoV 229E-based vaccine vectors can become a new class of highly efficient vaccines. First, the receptor of HCoV 229E, human aminopeptidase N (hAPN or CD13) is expressed mainly on human dendritic cells (DCs) and macrophages indicating that targeting of HCoV 229E-based vectors to professional antigen presenting cells can be achieved by receptor-mediated transduction. Second, HCoV 229E structural genes can be replaced by multiple transcriptional units encoding various antigens. These virus-like particles (VLPs) containing HCoV 229E-based vector RNA have the ability to transduce human DCs and to mediate heterologous gene expression in these cells. Finally, coronavirus infections are associated with mainly respiratory and enteric diseases, and natural transmission of coronaviruses occurs via mucosal surfaces. In humans, HCoV 229E causes common cold by infecting the upper respiratory tract. HCoV 229E infections are mainly encountered in children and re-infection occurs frequently in adults. It is thus most likely that pre-existing immunity against HCoV 229E will not significantly impact on the vaccination efficiency if HCoV 229E-based vectors are used in humans.

"Importantly, amino acid residues in all the 4 inserts have identity or similarity to those in the HIV-1 gp120 or HIV-1 Gag. Interestingly, despite the inserts being discontinuous on the primary amino acid sequence, 3D-modelling of the 2019-nCoV suggests that they converge to constitute the receptor binding site. The finding of 4 unique inserts in the 2019-nCoV, all of which have identity /similarity to amino acid residues in key structural proteins of HIV-1 is unlikely to be fortuitous in nature."
It would be a total screw-up to mix up a common cold virus and a novel bat virus.Leave a comment:
-
posted with permission
hat tip Jason Gale
Chinese Ambassador to U.S. Urges ‘Serious Rethinking’ of Ties
2020-04-22 06:09:29.947 GMT
By Bloomberg News
(Bloomberg) -- The Chinese ambassador to the U.S. called
for a “serious rethinking” of relations between the world’s
biggest economies in the face of the global coronavirus
pandemic.
“I think I should be hoping for more than just a pause in
tensions, but really a serious rethinking of the very
foundations of this important relationship,” Cui Tiankai said in
response to a question on U.S.-China ties during a Bloomberg New
Economy webcast on Tuesday.
Relations between the countries have continued to
deteriorate even after President Donald Trump and his Chinese
counterpart Xi Jinping agreed last month in a phone call to dial
down hostilities.
The U.S. leader, who is facing re-election this year, again
fanned speculation about the origins of the virus last weekend
when he said China should face consequences if it was “knowingly
responsible” for the outbreak. Missouri this week sued the
Chinese government for what it alleged was covering up the
extent of the coronavirus epidemic. China has denied those
claims, saying it has been transparent about the virus all
along.
Cui defended China’s record on transparency, saying the
country has shared all information it has with the World Health
Organization. He went on to criticize some U.S. politicians for
not paying enough attention to views from scientists, instead
being “preoccupied in their efforts for stigmatization and
groundless accusation.” He added that he’s concerned with the
“anything but China” mindset, in response to criticism that
Beijing has used medical donations for geopolitical purposes.
‘Shared Vulnerability’
Cui also rejected the idea that U.S.-China ties should be
defined as a strategic rivalry, particularly as the countries
are now faced with “shared vulnerability” amid the virus.
“Hopefully this pandemic will teach all of us a good lesson,” he
said, adding that the relationship should “be based on a more
realistic, forward-looking foundation.”
Cui said that China has done its best to achieve trust with
the U.S., and stands for dialogue as more discussion is needed.
“Fortunately at the top level, there has been a good working
relationship between the two presidents,” Cui said. “But of
course we have to do more at lower levels.”
Cui has emerged as a voice of caution on U.S.-China
relations. In March, he distanced himself from tweets by foreign
ministry spokesman Zhao Lijian, who speculated that the U.S.
Army may have brought the virus to Wuhan. Cui described such
speculation as “very harmful” and said investigations of the
virus’s origin were best left to scientists. Zhao has since
stopped promoting such claims.
Rare Spat Between Chinese Diplomats Signals Split Over
Trump
Cui added that China’s development has not come at the
expense of the U.S., saying China wants “nothing to do with U.S.
domestic politics, we can’t even make sense of it,” in response
to a question on who China would prefer to win the presidential
election in November.
“American people are most concerned about their own daily
lives, and don’t want to make China the centerpiece of their
domestic political debate,” he said.
To contact Bloomberg News staff for this story:
Sharon Chen in Beijing at schen462@bloomberg.net;
Jing Li in Beijing at jli1638@bloomberg.net;
Peter Martin in Beijing at pmartin138@bloomberg.net
To contact the editors responsible for this story:
Brendan Scott at bscott66@bloomberg.net
Jodi Schneider, Daniel Ten Kate
Leave a comment:
-
Translation Google
EXCLUSIVE - The thesis of a manipulated virus escaped from a Chinese laboratory: the pavement in the pool of Pr Luc Montagnier
By Th. B.
...
Posted on 04/16/2020 at 6:00 p.m. | |
THE ESSENTIAL
Professor Luc Montagnier, 2008 Nobel Prize laureate, says that SARS-CoV-2 is a manipulated virus accidentally released from a laboratory in Wuhan, China
Chinese researchers reportedly used this coronavirus as part of work to develop an AIDS vaccine
HIV DNA fragments allegedly found in the SARS-CoV-2 genome
We knew the Chinese version of the emergence of the coronavirus more and more undermined, but here is a thesis that tells a whole different story about the Covid-19 pandemic already responsible for more than 120,000 deaths in the world. According to Professor Luc Montagnier, 2008 Nobel Prize in Medicine for having "co-discovered" the HIV causing the AIDS epidemic with Fran?ois Barr?-Sinoussi today affirmed that SARS-CoV-2 is a manipulated virus and accidentally left a laboratory in Wuhan, China during the last quarter of 2019. This laboratory known to work on coronaviruses, according to Professor Montagnier, sought to use one of these viruses as an HIV vector as part of the looking for an AIDS vaccine!
"With my colleague, the bio-mathematician Jean-Claude Perez, we looked closely at the description of the genome of this RNA virus," explained Luc Montagnier, interviewed by Dr. Jean-Fran?ois Lemoine for the daily audio journal of Pourquoi Doctor , adding that others had already explored this track: "Indian researchers had already tried to publish the results of analyzes showing that this genome harbored sequences of another virus which is ... HIV, the virus of AIDS, but they were forced to retract, the pressures were too strong! ".
"To insert an HIV sequence into this genome, you need molecular tools"
Faced with these assertions by a professor who is sometimes challenged following iconoclastic positions, in particular on vaccination, one might also think that these conclusions are due to chance and that the coronavirus examined could have been taken from a patient otherwise afflicted with HIV. "No, replies Luc Montagnier, to insert an HIV sequence into this genome, molecular tools are needed, this can only be done in the laboratory".
According to the 2008 Nobel Prize in Medicine, the explanation is due to an "industrial accident" at the Wuhan laboratory. "The history of the fish market is a beautiful legend ... The assumption is that this virus left the laboratory because it escaped its promoters, it is a work of apprentice-sorcerer!", Estimates he promoting the thesis that the object of this work was the search for an AIDS vaccine.
"The truth always ends up breaking out"
This thesis defended by Professor Luc Montagnier generates in any case "reassuring" information. According to him, the altered elements of this virus eliminate themselves as it spreads: "Nature does not admit any molecular construction, it eliminates these foreign bodies ... even if nothing is done, things will get better, but after many deaths ...", he announces by advancing a solution. To stop the pandemic, Luc Montagnier says that by using "interfering waves, we could eliminate these sequences".
This is enough to fuel famous debates! To the point that Professor Montagnier's assertions could also classify him in the category of "conspirators": "Conspirators, it's the opposite camp, the one who hides the truth", he replies without wanting to accuse anyone but hoping that the Chinese would recognize what he said happened in their laboratory. "Anyway, the truth always ends up coming out, it's up to the Chinese government to take responsibility."
Leave a comment:
-
TAIPEI (Taiwan News) — Photos have resurfaced from 2018 showing a warped seal on a freezer door as suspicions grow that the Wuhan coronavirus (COVID-19) pandemic could have started at a poorly managed virology lab in Wuhan.
The official Chinese government explanation of the coronavirus pandemic is that it began in the city of Wuhan at a wet market known as the Huanan Seafood Wholesale Market. However, another theory to emerge recently is that the virus first made the jump to humans in one of two labs in Wuhan, where bat viruses were being studied.
Photos posted by the China Daily in early March, before later being scrubbed, show a seal around a freezer door looking dangerously warped. The original tweet proudly proclaimed the photos as being of the Wuhan Institute of Virology, where it said 1,500 viruses are kept.
On March 10, Twitter user John Pollizzi reposted one of the photos showing a badly bent seal and quipped: "I have seen better seals on my refrigerator in my kitchen." The image went viral, as many other netizens were equally horrified by the apparently lax safety measures, and was published on British media on Sunday (April 19). -
Photo reveals warped seal on Wuhan lab freezer door
China Daily photo of poor sealing goes viral as suspicions mount over safety measures at Wuhan virology lab
There is a very clear photo attached to this article.
Leave a comment:
-
I copied these posts from another thread today on April 17, 2020 and the vbulletin software placed them here in the thread because of the original date of the study post which precedes the date of this thread by 7 years. -
A coincidence I am sure:
China - Cangshan Nature Reserve in Yangbi County, Yunnan Province will be fully closed from March 29 until further notice
April 1st, 2020, 06:22 PM
https://flutrackers.com/forum/forum/...further-notice -
A coincidence I am sure:
China - Cangshan Nature Reserve in Yangbi County, Yunnan Province will be fully closed from March 29 until further notice
April 1st, 2020, 06:22 PM
https://flutrackers.com/forum/forum/...further-notice -
The rest of the paper. Link above. I am claiming fair use due to the pertinence under the current laws of the United States. This is not a commercial use.- Letter
- Published: 30 October 2013
- Xing-Yi Ge,
- Jia-Lu Li,
- Xing-Lou Yang,
- Aleksei A. Chmura,
- Guangjian Zhu,
- Jonathan H. Epstein,
- Jonna K. Mazet,
- Ben Hu,
- Wei Zhang,
- Cheng Peng,
- Yu-Ji Zhang,
- Chu-Ming Luo,
- Bing Tan,
- Ning Wang,
- Yan Zhu,
- Gary Crameri,
- Shu-Yi Zhang,
- Lin-Fa Wang,
- Peter Daszak &
- Zheng-Li Shi
Nature volume 503, pages535–538(2013)Cite this article- 82k Accesses
- 327 Citations
- 1349 Altmetric
- Metricsdetails
The 2002–3 pandemic caused by severe acute respiratory syndrome coronavirus (SARS-CoV) was one of the most significant public health events in recent history1. An ongoing outbreak of Middle East respiratory syndrome coronavirus2 suggests that this group of viruses remains a key threat and that their distribution is wider than previously recognized. Although bats have been suggested to be the natural reservoirs of both viruses3,4,5, attempts to isolate the progenitor virus of SARS-CoV from bats have been unsuccessful. Diverse SARS-like coronaviruses (SL-CoVs) have now been reported from bats in China, Europe and Africa5,6,7,8, but none is considered a direct progenitor of SARS-CoV because of their phylogenetic disparity from this virus and the inability of their spike proteins to use the SARS-CoV cellular receptor molecule, the human angiotensin converting enzyme II (ACE2)9,10.
Here we report whole-genome sequences of two novel bat coronaviruses from Chinese horseshoe bats (family: Rhinolophidae) in Yunnan, China: RsSHC014 and Rs3367. These viruses are far more closely related to SARS-CoV than any previously identified bat coronaviruses, particularly in the receptor binding domain of the spike protein. Most importantly, we report the first recorded isolation of a live SL-CoV (bat SL-CoV-WIV1) from bat faecal samples in Vero E6 cells, which has typical coronavirus morphology, 99.9% sequence identity to Rs3367 and uses ACE2 from humans, civets and Chinese horseshoe bats for cell entry.
Preliminary in vitro testing indicates that WIV1 also has a broad species tropism. Our results provide the strongest evidence to date that Chinese horseshoe bats are natural reservoirs of SARS-CoV, and that intermediate hosts may not be necessary for direct human infection by some bat SL-CoVs.
They also highlight the importance of pathogen-discovery programs targeting high-risk wildlife groups in emerging disease hotspots as a strategy for pandemic preparedness.
Main
The 2002–3 pandemic of SARS1 and the ongoing emergence of the Middle East respiratory syndrome coronavirus (MERS-CoV)2 demonstrate that CoVs are a significant public health threat. SARS-CoV was shown to use the human ACE2 molecule as its entry receptor, and this is considered a hallmark of its cross-species transmissibility11. The receptor binding domain (RBD) located in the amino-terminal region (amino acids 318–510) of the SARS-CoV spike (S) protein is directly involved in binding to ACE2 (ref. 12). However, despite phylogenetic evidence that SARS-CoV evolved from bat SL-CoVs, all previously identified SL-CoVs have major sequence differences from SARS-CoV in the RBD of their S proteins, including one or two deletions6,9. Replacing the RBD of one SL-CoV S protein with SARS-CoV S conferred the ability to use human ACE2 and replicate efficiently in mice9,13. However, to date, no SL-CoVs have been isolated from bats, and no wild-type SL-CoV of bat origin has been shown to use ACE2.
We conducted a 12-month longitudinal survey (April 2011–September 2012) of SL-CoVs in a colony of Rhinolophus sinicus at a single location in Kunming, Yunnan Province, China (Extended Data Table 1). A total of 117 anal swabs or faecal samples were collected from individual bats using a previously published method5,14. A one-step reverse transcription (RT)-nested PCR was conducted to amplify the RNA-dependent RNA polymerase (RdRP) motifs A and C, which are conserved among alphacoronaviruses and betacoronaviruses15.
Twenty-seven of the 117 samples (23%) were classed as positive by PCR and subsequently confirmed by sequencing. The species origin of all positive samples was confirmed to be R. sinicus by cytochrome b sequence analysis, as described previously16. A higher prevalence was observed in samples collected in October (30% in 2011 and 48.7% in 2012) than those in April (7.1% in 2011) or May (7.4% in 2012) (Extended Data Table 1). Analysis of the S protein RBD sequences indicated the presence of seven different strains of SL-CoVs (Fig. 1a and Extended Data Figs 1 and 2). In addition to RBD sequences, which closely matched previously described SL-CoVs (Rs672, Rf1 and HKU3)5,8,17,18, two novel strains (designated SL-CoV RsSHC014 and Rs3367) were discovered. Their full-length genome sequences were determined, and both were found to be 29,787 base pairs in size (excluding the poly(A) tail). The overall nucleotide sequence identity of these two genomes with human SARS-CoV (Tor2 strain) is 95%, higher than that observed previously for bat SL-CoVs in China (88–92%)5,8,17,18 or Europe (76%)6 (Extended Data Table 2 and Extended Data Figs 3 and 4). Higher sequence identities were observed at the protein level between these new SL-CoVs and SARS-CoVs (Extended Data Tables 3 and 4). To understand the evolutionary origin of these two novel SL-CoV strains, we conducted recombination analysis with the Recombination Detection Program 4.0 package19 using available genome sequences of bat SL-CoV strains (Rf1, Rp3, Rs672, Rm1, HKU3 and BM48-31) and human and civet representative SARS-CoV strains (BJ01, SZ3, Tor2 and GZ02). Three breakpoints were detected with strong P values (<10−20) and supported by similarity plot and bootscan analysis (Extended Data Fig. 5a, b). Breakpoints were located at nucleotides 20,827, 26,553 and 28,685 in the Rs3367 (and RsSHC014) genome, and generated recombination fragments covering nucleotides 20,827–26,533 (5,727 nucleotides) (including partial open reading frame (ORF) 1b, full-length S, ORF3, E and partial M gene) and nucleotides 26,534–28,685 (2,133 nucleotides) (including partial ORF M, full-length ORF6, ORF7, ORF8 and partial N gene). Phylogenetic analysis using the major and minor parental regions suggested that Rs3367, or RsSHC014, is the descendent of a recombination of lineages that ultimately lead to SARS-CoV and SL-CoV Rs672 (Fig. 1b).
Figure 1: Phylogenetic tree based on amino acid sequences of the S RBD region and the two parental regions of bat SL-CoV Rs3367 or RsSHC014.
a, SARS-CoV S protein amino acid residues 310–520 were aligned with homologous regions of bat SL-CoVs using the ClustalW software. A maximum-likelihood phylogenetic tree was constructed using a Poisson model with bootstrap values determined by 1,000 replicates in the MEGA5 software package. The RBD sequences identified in this study are in bold and named by the sample numbers. The key amino acid residues involved in interacting with the human ACE2 molecule are indicated on the right of the tree. SARS-CoV GZ02, BJ01 and Tor2 were isolated from patients in the early, middle and late phase, respectively, of the SARS outbreak in 2003. SARS-CoV SZ3 was identified from Paguma larvata in 2003 collected in Guangdong, China. SL-CoV Rp3, Rs672 and HKU3-1 were identified from R. sinicus collected in China (respectively: Guangxi, 2004; Guizhou, 2006; Hong Kong, 2005). Rf1 and Rm1 were identified from R. ferrumequinum and R. macrotis, respectively, collected in Hubei, China, in 2004. Bat SARS-related CoV BM48-31 was identified from R. blasii collected in Bulgaria in 2008. Bat CoV HKU9-1 was identified from Rousettus leschenaultii collected in Guangdong, China in 2005/2006 and used as an outgroup. All sequences in bold and italics were identified in the current study. Filled triangles, circles and diamonds indicate samples with co-infection by two different SL-CoVs. ‘–’ indicates the amino acid deletion. b, Phylogenetic origins of the two parental regions of Rs3367 or RsSHC014. Maximum likelihood phylogenetic trees were constructed from alignments of two fragments covering nucleotides 20,827–26,533 (5,727 nucleotides) and 26,534 –28,685 (2,133 nucleotides) of the Rs3367 genome, respectively. For display purposes, the trees were midpoint rooted. The taxa were annotated according to strain names: SARS-CoV, SARS coronavirus; SARS-like CoV, bat SARS-like coronavirus. The two novel SL-CoVs, Rs3367 and RsSHC014, are in bold and italics.
PowerPoint slide
Full size image
The most notable sequence differences between these two new SL-CoVs and previously identified SL-CoVs is in the RBD regions of their S proteins. First, they have higher amino acid sequence identity to SARS-CoV (85% and 96% for RsSHC014 and Rs3367, respectively). Second, there are no deletions and they have perfect sequence alignment with the SARS-CoV RBD region (Extended Data Figs 1 and 2). Structural and mutagenesis studies have previously identified five key residues (amino acids 442, 472, 479, 487 and 491) in the RBD of the SARS-CoV S protein that have a pivotal role in receptor binding20,21. Although all five residues in the RsSHC014 S protein were found to be different from those of SARS-CoV, two of the five residues in the Rs3367 RBD were conserved (Fig. 1 and Extended Data Fig. 1).
Despite the rapid accumulation of bat CoV sequences in the last decade, there has been no report of successful virus isolation6,22,23. We attempted isolation from SL-CoV PCR-positive samples. Using an optimized protocol and Vero E6 cells, we obtained one isolate which caused cytopathic effect during the second blind passage. Purified virions displayed typical coronavirus morphology under electron microscopy (Fig. 2). Sequence analysis using a sequence-independent amplification method14 to avoid PCR-introduced contamination indicated that the isolate was almost identical to Rs3367, with 99.9% nucleotide genome sequence identity and 100% amino acid sequence identity for the S1 region. The new isolate was named SL-CoV-WIV1.
Figure 2: Electron micrograph of purified virions.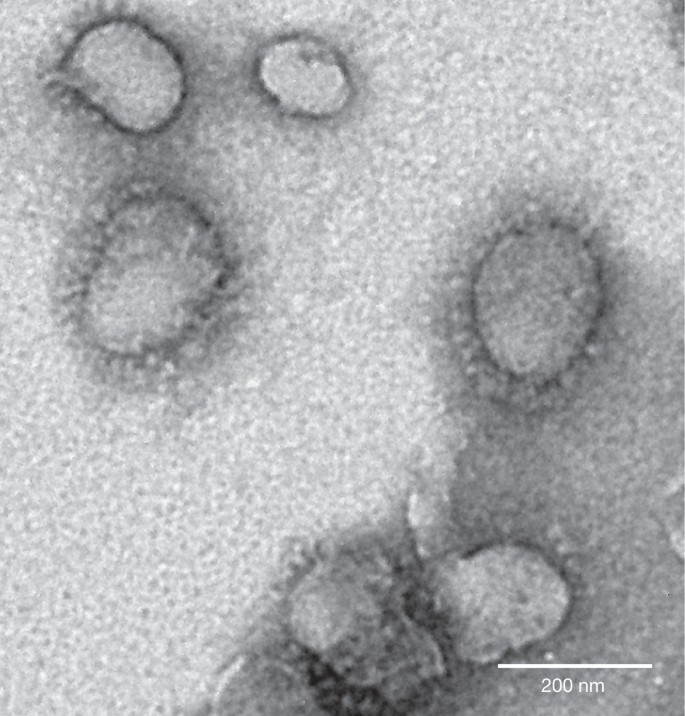
Virions from a 10-ml culture were collected, fixed and concentrated/purified by sucrose gradient centrifugation. The pelleted viral particles were suspended in 100 μl PBS, stained with 2% phosphotungstic acid (pH 7.0) and examined directly using a Tecnai transmission electron microscope (FEI) at 200 kV.
PowerPoint slide
Full size image
To determine whether WIV1 can use ACE2 as a cellular entry receptor, we conducted virus infectivity studies using HeLa cells expressing or not expressing ACE2 from humans, civets or Chinese horseshoe bats. We found that WIV1 is able to use ACE2 of different origins as an entry receptor and replicated efficiently in the ACE2-expressing cells (Fig. 3). This is, to our knowledge, the first identification of a wild-type bat SL-CoV capable of using ACE2 as an entry receptor.
Figure 3: Analysis of receptor usage of SL-CoV-WIV1 determined by immunofluorescence assay and real-time PCR.
Determination of virus infectivity in HeLa cells with and without the expression of ACE2. b, bat; c, civet; h, human. ACE2 expression was detected with goat anti-humanACE2 antibody followed by fluorescein isothiocyanate (FITC)-conjugated donkey anti-goat IgG. Virus replication was detected with rabbit antibody against the SL-CoV Rp3 nucleocapsid protein followed by cyanine 3 (Cy3)-conjugated mouse anti-rabbit IgG. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). The columns (from left to right) show staining of nuclei (blue), ACE2 expression (green), virus replication (red), merged triple-stained images and real-time PCR results, respectively. (n = 3); error bars represent standard deviation.
PowerPoint slide
Full size image
To assess its cross-species transmission potential, we conducted infectivity assays in cell lines from a range of species. Our results (Fig. 4 and Extended Data Table 5) indicate that bat SL-CoV-WIV1 can grow in human alveolar basal epithelial (A549), pig kidney 15 (PK-15) and Rhinolophus sinicus kidney (RSKT) cell lines, but not in human cervix (HeLa), Syrian golden hamster kidney (BHK21), Myotis davidii kidney (BK), Myotis chinensis kidney (MCKT), Rousettus leschenaulti kidney (RLK) or Pteropus alecto kidney (PaKi) cell lines. Real-time RT–PCR indicated that WIV1 replicated much less efficiently in A549, PK-15 and RSKT cells than in Vero E6 cells (Fig. 4).
Figure 4: Analysis of host range of SL-CoV-WIV1 determined by immunofluorescence assay and real-time PCR.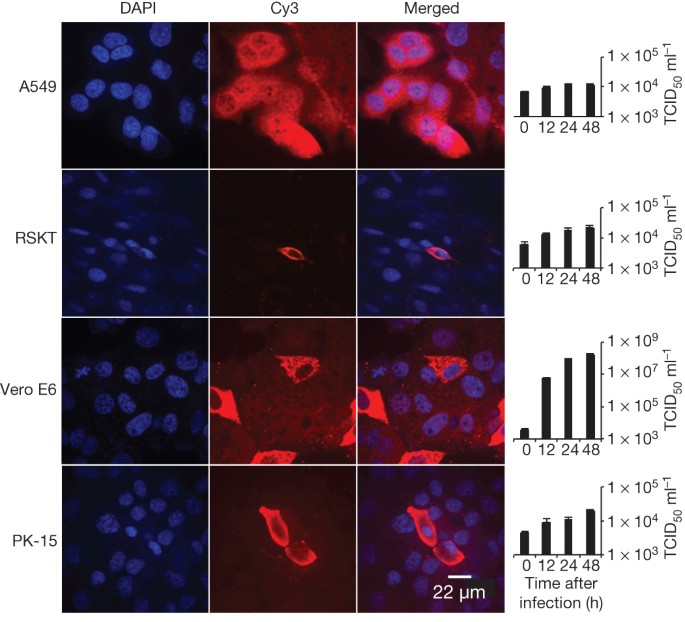
Virus infection in A549, RSKT, Vero E6 and PK-15 cells. Virus replication was detected as described for Fig. 3. The columns (from left to right) show staining of nuclei (blue), virus replication (red), merged double-stained images and real-time PCR results, respectively. n = 3; error bars represent s.d.
PowerPoint slide
Full size image
To assess the cross-neutralization activity of human SARS-CoV sera against WIV1, we conducted serum-neutralization assays using nine convalescent sera from SARS patients collected in 2003. The results showed that seven of these were able to completely neutralize 100 tissue culture infectious dose 50 (TCID50) WIV1 at dilutions of 1:10 to 1:40, further confirming the close relationship between WIV1 and SARS-CoV.
Our findings have important implications for public health. First, they provide the clearest evidence yet that SARS-CoV originated in bats. Our previous work provided phylogenetic evidence of this5, but the lack of an isolate or evidence that bat SL-CoVs can naturally infect human cells, until now, had cast doubt on this hypothesis. Second, the lack of capacity of SL-CoVs to use of ACE2 receptors has previously been considered as the key barrier for their direct spillover into humans, supporting the suggestion that civets were intermediate hosts for SARS-CoV adaptation to human transmission during the SARS outbreak24. However, the ability of SL-CoV-WIV1 to use human ACE2 argues against the necessity of this step for SL-CoV-WIV1 and suggests that direct bat-to-human infection is a plausible scenario for some bat SL-CoVs. This has implications for public health control measures in the face of potential spillover of a diverse and growing pool of recently discovered SARS-like CoVs with a wide geographic distribution.
Our findings suggest that the diversity of bat CoVs is substantially higher than that previously reported. In this study we were able to demonstrate the circulation of at least seven different strains of SL-CoVs within a single colony of R. sinicus during a 12-month period. The high genetic diversity of SL-CoVs within this colony was mirrored by high phenotypic diversity in the differential use of ACE2 by different strains. It would therefore not be surprising if further surveillance reveals a broad diversity of bat SL-CoVs that are able to use ACE2, some of which may have even closer homology to SARS-CoV than SL-CoV-WIV1. Our results—in addition to the recent demonstration of MERS-CoV in a Saudi Arabian bat25, and of bat CoVs closely related to MERS-CoV in China, Africa, Europe and North America3,26,27—suggest that bat coronaviruses remain a substantial global threat to public health.
Finally, this study demonstrates the public health importance of pathogen discovery programs targeting wildlife that aim to identify the ‘known unknowns’—previously unknown viral strains closely related to known pathogens. These programs, focused on specific high-risk wildlife groups and hotspots of disease emergence, may be a critical part of future global strategies to predict, prepare for, and prevent pandemic emergence28.
Methods Summary
Throat and faecal swabs or fresh faecal samples were collected in viral transport medium as described previously14. All PCR was conducted with the One-Step RT–PCR kit (Invitrogen). Primers targeting the highly conserved regions of the RdRP gene were used for detection of all alphacoronaviruses and betacoronaviruses as described previously15. Degenerate primers were designed on the basis of all available genomic sequences of SARS-CoVs and SL-CoVs and used for amplification of the RBD sequences of S genes or full-length genomic sequences. Degenerate primers were used for amplification of the bat ACE2 gene as described previously29. PCR products were gel purified and cloned into pGEM-T Easy Vector (Promega). At least four independent clones were sequenced to obtain a consensus sequence. PCR-positive faecal samples (in 200 μl buffer) were gradient centrifuged at 3,000–12,000g and supernatant diluted at 1:10 in DMEM before being added to Vero E6 cells. After incubation at 37 ?C for 1 h, inocula were removed and replaced with fresh DMEM with 2% FCS. Cells were incubated at 37 ?C and checked daily for cytopathic effect. Cell lines from different origins were grown on coverslips in 24-well plates and inoculated with the novel SL-CoV at a multiplicity of infection of 10. Virus replication was detected at 24 h after infection using rabbit antibodies against the SL-CoV Rp3 nucleocapsid protein followed by Cy3-conjugated goat anti-rabbit IgG.
Online Methods
Sampling
Bats were trapped in their natural habitat as described previously5. Throat and faecal swab samples were collected in viral transport medium (VTM) composed of Hank’s balanced salt solution, pH 7.4, containing BSA (1%), amphotericin (15 μg ml−1), penicillin G (100 U ml−1) and streptomycin (50 μg ml−1). To collect fresh faecal samples, clean plastic sheets measuring 2.0 by 2.0 m were placed under known bat roosting sites at about 18:00 h each evening. Relatively fresh faecal samples were collected from sheets at approximately 05:30–06:00 the next morning and placed in VTM. Samples were transported to the laboratory and stored at −80 ?C until use. All animals trapped for this study were released back to their habitat after sample collection. All sampling processes were performed by veterinarians with approval from Animal Ethics Committee of the Wuhan Institute of Virology (WIVH05210201) and EcoHealth Alliance under an inter-institutional agreement with University of California, Davis (UC Davis protocol no. 16048).
RNA extraction, PCR and sequencing
RNA was extracted from 140 μl of swab or faecal samples with a Viral RNA Mini Kit (Qiagen) following the manufacturer’s instructions. RNA was eluted in 60 μl RNAse-free buffer (buffer AVE, Qiagen), then aliquoted and stored at −80 ?C. One-step RT–PCR (Invitrogen) was used to detect coronavirus sequences as described previously15. First round PCR was conducted in a 25-μl reaction mix containing 12.5 μl PCR 2? reaction mix buffer, 10 pmol of each primer, 2.5 mM MgSO4, 20 U RNase inhibitor, 1 μl SuperScript III/ Platinum Taq Enzyme Mix and 5 μl RNA. Amplification of the RdRP-gene fragment was performed as follows: 50 ?C for 30 min, 94 ?C for 2 min, followed by 40 cycles consisting of 94 ?C for 15 s, 62 ?C for 15 s, 68 ?C for 40 s, and a final extension of 68 ?C for 5 min. Second round PCR was conducted in a 25-μl reaction mix containing 2.5 μl PCR reaction buffer, 5 pmol of each primer, 50 mM MgCl2, 0.5 mM dNTP, 0.1 μl Platinum Taq Enzyme (Invitrogen) and 1 μl first round PCR product. The amplification of RdRP-gene fragment was performed as follows: 94 ?C for 5 min followed by 35 cycles consisting of 94 ?C for 30 s, 52 ?C for 30 s, 72 ?C for 40 s, and a final extension of 72 ?C for 5 min.
To amplify the RBD region, one-step RT–PCR was performed with primers designed based on available SARS-CoV or bat SL-CoVs (first round PCR primers; F, forward; R, reverse: CoVS931F-5′-VWGADGTTGTKAGRTTYCCT-3′ and CoVS1909R-5′-TAARACAVCCWGCYTGWGT-3′; second PCR primers: CoVS951F-5′-TGTKAGRTTYCCTAAYATTAC-3′ and CoVS1805R-5′-ACATCYTGATANARAACAGC-3′). First-round PCR was conducted in a 25-μl reaction mix as described above except primers specific for the S gene were used. The amplification of the RBD region of the S gene was performed as follows: 50 ?C for 30 min, 94 ?C for 2 min, followed by 35 cycles consisting of 94 ?C for 15 s, 43 ?C for 15 s, 68 ?C for 90 s, and a final extension of 68 ?C for 5 min. Second-round PCR was conducted in a 25-μl reaction mix containing 2.5 μl PCR reaction buffer, 5 pmol of each primer, 50 mM MgCl2, 0.5 mM dNTP, 0.1 μl Platinum Taq Enzyme (Invitrogen) and 1 μl first round PCR product. Amplification was performed as follows: 94 ?C for 5 min followed by 40 cycles consisting of 94 ?C for 30 s, 41 ?C for 30 s, 72 ?C for 60 s, and a final extension of 72 ?C for 5 min.
PCR products were gel purified and cloned into pGEM-T Easy Vector (Promega). At least four independent clones were sequenced to obtain a consensus sequence for each of the amplified regions.
Sequencing full-length genomes
Degenerate coronavirus primers were designed based on all available SARS-CoV and bat SL-CoV sequences in GenBank and specific primers were designed from genome sequences generated from previous rounds of sequencing in this study (primer sequences will be provided upon request). All PCRs were conducted using the One-Step RT–PCR kit (Invitrogen). The 5′ and 3′ genomic ends were determined using the 5′ or 3′ RACE kit (Roche), respectively. PCR products were gel purified and sequenced directly or following cloning into pGEM-T Easy Vector (Promega). At least four independent clones were sequenced to obtain a consensus sequence for each of the amplified regions and each region was sequenced at least twice.
Sequence analysis and databank accession numbers
Routine sequence management and analysis was carried out using DNAStar or Geneious. Sequence alignment and editing was conducted using ClustalW, BioEdit or GeneDoc. Maximum Likelihood phylogenetic trees based on the protein sequences were constructed using a Poisson model with bootstrap values determined by 1,000 replicates in the MEGA5 software package.
Sequences obtained in this study have been deposited in GenBank as follows (accession numbers given in parenthesis): full-length genome sequence of SL-CoV RsSHC014 and Rs3367 (KC881005, KC881006); full-length sequence of WIV1 S (KC881007); RBD (KC880984-KC881003); ACE2 (KC8810040). SARS-CoV sequences used in this study: human SARS-CoV strains Tor2 (AY274119), BJ01 (AY278488), GZ02 (AY390556) and civet SARS-CoV strain SZ3 (AY304486). Bat coronavirus sequences used in this study: Rs672 (FJ588686), Rp3 (DQ071615), Rf1 (DQ412042), Rm1 (DQ412043), HKU3-1 (DQ022305), BM48-31 (NC_014470), HKU9-1 (NC_009021), HKU4 (NC_009019), HKU5 (NC_009020), HKU8 (DQ249228), HKU2 (EF203067), BtCoV512 (NC_009657), 1A (NC_010437). Other coronavirus sequences used in this study: HCoV-229E (AF304460), HCoV-OC43 (AY391777), HCoV-NL63 (AY567487), HKU1 (NC_006577), EMC (JX869059), FIPV (NC_002306), PRCV (DQ811787), BWCoV (NC_010646), MHV (AY700211), IBV (AY851295).
Amplification, cloning and expression of the bat ACE2 gene
Construction of expression clones for human and civet ACE2 in pcDNA3.1 has been described previously29. Bat ACE2 was amplified from a R. sinicus (sample no. 3357). In brief, total RNA was extracted from bat rectal tissue using the RNeasy Mini Kit (Qiagen). First-strand complementary DNA was synthesized from total RNA by reverse transcription with random hexamers. Full-length bat ACE2 fragments were amplified using forward primer bAF2 and reverse primer bAR2 (ref. 29). The ACE2 gene was cloned into pCDNA3.1 with KpnI and XhoI, and verified by sequencing. Purified ACE2 plasmids were transfected to HeLa cells. After 24 h, lysates of HeLa cells expressing human, civet, or bat ACE2 were confirmed by western blot or immunofluorescence assay.
Western blot analysis
Lysates of cells or filtered supernatants containing pseudoviruses were separated by SDS–PAGE, followed by transfer to a nitrocellulose membrane (Millipore). For detection of S protein, the membrane was incubated with rabbit anti-Rp3 S fragment (amino acids 561–666) polyantibodies (1:200), and the bound antibodies were detected by alkaline phosphatase (AP)-conjugated goat anti-rabbit IgG (1:1,000). For detection of HIV-1 p24 in supernatants, monoclonal antibody against HIV p24 (p24 MAb) was used as the primary antibody at a dilution of 1:1,000, followed by incubation with AP-conjugated goat anti-mouse IgG at the same dilution. To detect the expression of ACE2 in HeLa cells, goat antibody against the human ACE2 ectodomain (1:500) was used as the first antibody, followed by incubation with horseradish peroxidase-conjugated donkey anti-goat IgG (1:1,000).
Virus isolation
Vero E6 cell monolayers were maintained in DMEM supplemented with 10% FCS. PCR-positive samples (in 200 μl buffer) were gradient centrifuged at 3,000–12,000g, and supernatant were diluted 1:10 in DMEM before being added to Vero E6 cells. After incubation at 37 ?C for 1 h, inocula were removed and replaced with fresh DMEM with 2% FCS. Cells were incubated at 37 ?C for 3 days and checked daily for cytopathic effect. Double-dose triple antibiotics penicillin/streptomycin/amphotericin (Gibco) were included in all tissue culture media (penicillin 200 IU ml−1, streptomycin 0.2 mg ml−1, amphotericin 0.5 μg ml−1). Three blind passages were carried out for each sample. After each passage, both the culture supernatant and cell pellet were examined for presence of virus by RT–PCR using primers targeting the RdRP or S gene. Virions in supernatant (10 ml) were collected and fixed using 0.1% formaldehyde for 4 h, then concentrated by ultracentrifugation through a 20% sucrose cushion (5 ml) at 80,000g for 90 min using a Ty90 rotor (Beckman). The pelleted viral particles were suspended in 100 μl PBS, stained with 2% phosphotungstic acid (pH 7.0) and examined using a Tecnai transmission electron microscope (FEI) at 200 kV.
Virus infectivity detected by immunofluorescence assay
Cell lines used for this study and their culture conditions are summarized in Extended Data Table 5. Virus titre was determined in Vero E6 cells by cytopathic effect (CPE) counts. Cell lines from different origins and HeLa cells expressing ACE2 from human, civet or Chinese horseshoe bat were grown on coverslips in 24-well plates (Corning) incubated with bat SL-CoV-WIV1 at a multiplicity of infection = 10 for 1 h. The inoculum was removed and washed twice with PBS and supplemented with medium. HeLa cells without ACE2 expression and Vero E6 cells were used as negative and positive controls, respectively. At 24 h after infection, cells were washed with PBS and fixed with 4% formaldehyde in PBS (pH 7.4) for 20 min at 4 ?C. ACE2 expression was detected using goat anti-human ACE2 immunoglobulin (R&D Systems) followed by FITC-labelled donkey anti-goat immunoglobulin (PTGLab). Virus replication was detected using rabbit antibody against the SL-CoV Rp3 nucleocapsid protein followed by Cy3-conjugated mouse anti-rabbit IgG. Nuclei were stained with DAPI. Staining patterns were examined using a FV1200 confocal microscope (Olympus).
Virus infectivity detected by real-time RT–PCR
Vero E6, A549, PK15, RSKT and HeLa cells with or without expression of ACE2 of different origins were inoculated with 0.1 TCID50 WIV-1 and incubated for 1 h at 37 ?C. After removing the inoculum, the cells were cultured with medium containing 1% FBS. Supernatants were collected at 0, 12, 24 and 48 h. RNA from 140 μl of each supernatant was extracted with the Viral RNA Mini Kit (Qiagen) following manufacturer’s instructions and eluted in 60 μl buffer AVE (Qiagen). RNA was quantified on the ABI StepOne system, with the TaqMan AgPath-ID One-Step RT–PCR Kit (Applied Biosystems) in a 25 μl reaction mix containing 4 μl RNA, 1 ? RT–PCR enzyme mix, 1 ? RT–PCR buffer, 40 pmol forward primer (5′-GTGGTGGTGACGGCAAAATG-3′), 40 pmol reverse primer (5′-AAGTGAAGCTTCTGGGCCAG-3′) and 12 pmol probe (5′-FAM-AAAGAGCTCAGCCCCAGATG-BHQ1-3′). Amplification parameters were 10 min at 50 ?C, 10 min at 95 ?C and 50 cycles of 15 s at 95 ?C and 20 s at 60 ?C. RNA dilutions from purified WIV-1 stock were used as a standard.
Serum neutralization test
SARS patient sera were inactivated at 56 ?C for 30 min and then used for virus neutralization testing. Sera were diluted starting with 1:10 and then serially twofold diluted in 96-well cell plates to 1:40. Each 100 μl serum dilution was mixed with 100 μl viral supernatant containing 100 TCID50of WIV1 and incubated at 37 ?C for 1 h. The mixture was added in triplicate wells of 96-well cell plates with plated monolayers of Vero E6 cells and further incubated at 37 ?C for 2 days. Serum from a healthy blood donor was used as a negative control in each experiment. CPE was observed using an inverted microscope 2 days after inoculation. The neutralizing antibody titre was read as the highest dilution of serum which completely suppressed CPE in infected wells. The neutralization test was repeated twice.
Recombination analysis
Full-length genomic sequences of SL-CoV Rs3367 or RsSHC014 were aligned with those of selected SARS-CoVs and bat SL-CoVs using Clustal X. The aligned sequences were preliminarily scanned for recombination events using Recombination Detection Program (RDP) 4.0 (ref. 19). The potential recombination events suggested by RDP owing to their strong P values (<10–20) were investigated further by similarity plot and bootscan analyses implemented in Simplot 3.5.1. Phylogenetic origin of the major and minor parental regions of Rs3367 or RsSHC014 were constructed from the concatenated sequences of the essential ORFs of the major and minor parental regions of selected SARS-CoV and SL-CoVs. Two genome regions between three estimated breakpoints (20,827–26,553 and 26,554–28,685) were aligned independently using ClustalX and generated two alignments of 5,727 base pairs and 2,133 base pairs. The two alignments were used to construct maximum likelihood trees to better infer the fragment parents. All nucleotide numberings in this study are based on Rs3367 genome position.
Accession codes
Accessions
GenBank/EMBL/DDBJData deposits
Sequences of three bat SL-CoV genomes, bat SL-CoV RBD and R. sinicus ACE2 genes have been deposited in GenBank under accession numbers KC881005–KC881007 (genomes from SL-CoV RsSHC014, Rs3367 and W1V1, respectively), KC880984–KC881003 (bat SL-CoV RBD genes) and KC881004 (R. sinicus ACE2), respectively.
References- 1
Ksiazek, T. G. et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966 (2003) - 2
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. & Fouchier, R. A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820 (2012) - 3
Anthony, S. J. et al. Coronaviruses in bats from Mexico. J. Gen. Virol. 94, 1028–1038 (2013) - 4
Raj, V. S. et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495, 251–254 (2013) - 5
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005) - 6
Drexler, J. F. et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 84, 11336–11349 (2010) - 7
Tong, S. et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15, 482–485 (2009) - 8
Lau, S. K. P. et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl Acad. Sci. USA 102, 14040–14045 (2005) - 9
Ren, W. et al. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 82, 1899–1907 (2008) - 10
Hon, C. C. et al. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 82, 1819–1826 (2008) - 11
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003) - 12
Wong, S. K., Li, W., Moore, M. J., Choe, H. & Farzan, M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 279, 3197–3201 (2004) - 13
Becker, M. M. et al. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl Acad. Sci. USA 105, 19944–19949 (2008) - 14
Li, Y. et al. Host range, prevalence, and genetic diversity of adenoviruses in bats. J. Virol. 84, 3889–3897 (2010) - 15
De Souza Luna, L. K. et al. Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J. Clin. Microbiol. 45, 1049–1052 (2007) - 16
Cui, J. et al. Evolutionary relationships between bat coronaviruses and their hosts. Emerg. Infect. Dis. 13, 1526–1532 (2007) - 17
Yuan, J. et al. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 91, 1058–1062 (2010) - 18
Ren, W. et al. Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J. Gen. Virol. 87, 3355–3359 (2006) - 19
Martin, D. P. et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26, 2462–2463 (2010) - 20
Wu, K., Peng, G., Wilken, M., Geraghty, R. J. & Li, F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 287, 8904–8911 (2012) - 21
Li, W. et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 24, 1634–1643 (2005) - 22
Lau, S. K. et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 84, 2808–2819 (2010) - 23
Lau, S. K. et al. Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J. Virol. 84, 11385–11394 (2010) - 24
Song, H. D. et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl Acad. Sci. USA 102, 2430–2435 (2005) - 25
Memish, Z. A. et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 19, 11 (2013) - 26
Chan, J. F. et al. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 65, 477–489 (2012) - 27
Ithete, N. L. et al. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 19, 1697–1699 (2013) - 28
Morse, S. S. et al. Prediction and prevention of the next pandemic zoonosis. Lancet 380, 1956–1965 (2012) - 29
Hou, Y. et al. Angiotensin-converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS-CoV entry. Arch. Virol. 155, 1563–1569 (2010)
Download references
Acknowledgements
We acknowledge financial support from the State Key Program for Basic Research (2011CB504701 and 2010CB530100), National Natural Science Foundation of China (81290341 and 31321001), Scientific and technological basis special project (2013FY113500), CSIRO OCE Science Leaders Award, National Institute of Allergy and Infectious Diseases (NIAID) award number R01AI079231, a National Institutes of Health (NIH)/National Science Foundation (NSF) ‘Ecology and Evolution of Infectious Diseases’ award from the NIH Fogarty International Center (R01TW005869), an award from the NIH Fogarty International Center supported by International Influenza Funds from the Office of the Secretary of the Department of Health and Human Services (R56TW009502), and United States Agency for International Development (USAID) Emerging Pandemic Threats PREDICT. The contents are the responsibility of the authors and do not necessarily reflect the views of NIAID, NIH, NSF, USAID or the United States Government. We thank X. Che from Zhujiang Hospital, Southern Medical University, for providing human SARS patient sera.
Author information
Author notes- Xing-Yi Ge, Jia-Lu Li and Xing-Lou Yang: These authors contributed equally to this work.
- Center for Emerging Infectious Diseases, State Key Laboratory of Virology, Wuhan Institute of Virology of the Chinese Academy of Sciences, Wuhan, 430071, China
- Xing-Yi Ge
- , Jia-Lu Li
- , Xing-Lou Yang
- , Ben Hu
- , Wei Zhang
- , Cheng Peng
- , Yu-Ji Zhang
- , Chu-Ming Luo
- , Bing Tan
- , Ning Wang
- , Yan Zhu
- & Zheng-Li Shi
- EcoHealth Alliance, New York, 10001, New York, USA
- Aleksei A. Chmura
- , Guangjian Zhu
- , Jonathan H. Epstein
- & Peter Daszak
- One Health Institute, School of Veterinary Medicine, University of California, Davis, 95616, California, USA
- Jonna K. Mazet
- CSIRO Australian Animal Health Laboratory, Geelong, 3220, Victoria, Australia
- Gary Crameri
- & Lin-Fa Wang
- College of Life Sciences, East China Normal University, Shanghai 200062, China
- Shu-Yi Zhang
- Emerging Infectious Diseases Program, Duke-NUS Graduate Medical School, Singapore 169857
- Lin-Fa Wang
Z.-L.S. and P.D. designed and coordinated the study. X.-Y.G., J.-L. L. and X.-L.Y. conducted majority of experiments and contributed equally to the study. A.A.C., B.H., W.Z., C.P., Y.-J.Z., C.-M.L., B.T., N.W. and Y.Z. conducted parts of the experiments and analyses. J.H.E., J.K.M. and S.-Y.Z. coordinated the field study. X.-Y.G., J.-L.L., X.-L.Y., B.T. and G.-J.Z. collected the samples. G.C. and L.-F.W. designed and supervised part of the experiments. All authors contributed to the interpretations and conclusions presented. Z.-L.S. and X-Y.G. wrote the manuscript with significant contributions from P.D. and L-F.W. and input from all authors.
Corresponding authors
Correspondence to Peter Daszak or Zheng-Li Shi.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Sequence alignment of CoV S protein RBD.
SARS-CoV S protein (amino acids 310–520) is aligned with homologous regions of bat SL-CoVs using ClustalW. The newly discovered bat SL-CoVs are indicated with a bold vertical line on the left. The key amino acid residues involved in the interaction with human ACE2 are numbered on the top of the aligned sequences.
Extended Data Figure 2 Alignment of CoV S protein S1 sequences.
Alignment of S1 sequences (amino acids 1–660) of the two novel bat SL-CoV S proteins with those of previously reported bat SL-CoVs and human and civet SARS-CoVs. The newly discovered bat SL-CoVs are boxed in red. SARS-CoV GZ02, BJ01 and Tor2 were isolated from patients in the early, middle and late phase, respectively, of the SARS outbreak in 2003. SARS-CoV SZ3 was identified from P. larvata in 2003 collected in Guangdong, China. SL-CoV Rp3, Rs 672 and HKU3-1 were identified from R. sinicus collected in Guangxi, Guizhou and Hong Kong, China, respectively. Rf1 and Rm1 were identified from R. ferrumequinum and R. macrotis, respectively, collected in Hubei Province, China. Bat SARS-related CoV BM48-31 was identified from R. blasii collected in Bulgaria.
Extended Data Figure 3 Complete RdRP sequence phylogeny.
Phylogenetic tree of bat SL-CoVs and SARS-CoVs on the basis of complete RdRP sequences (2,796 nucleotides). Bat SL-CoVs RsSHC014 and Rs3367 are highlighted by filled circles. Three established coronaivirus genera, Alphacoronavirus, Betacoronavirus and Gammacoronavirus are marked as α, β and γ, respectively. Four CoV groups in the genus Betacoronavirus are indicated as A, B, C and D, respectively. MHV, murine hepatitis virus; PHEV, porcine haemagglutinating encephalomyelitis virus; PRCV, porcine respiratory coronavirus; FIPV, feline infectious peritonitis virus; IBV, infectious bronchitis coronavirus; BW, beluga whale coronavirus.
Extended Data Figure 4 Sequence phylogeny of the complete S protein of SL-CoVs and SARS-CoV.
Phylogenetic tree of bat SL-CoVs and SARS-CoVs on the basis of complete S protein sequences (1,256 amino acids). Bat SL-CoVs RsSHC014 and Rs3367 are highlighted by filled circles. Bat CoV HKU9 was used as an outgroup.
Extended Data Figure 5 Detection of potential recombination events.
a, b, Similarity plot (a) and bootscan analysis (b) detected three recombination breakpoints in the bat SL-CoV Rs3367 or SHC014 genome. The three breakpoints were located at the ORF1b (nt 20,827), M (nucleotides 26,553) and N (nucleotides 28,685) genes, respectively. Both analyses were performed with an F84 distance model, a window size of 1,500 base pairs and a step size of 300 base pairs.
Extended Data Table 1 Summary of sampling detail and CoV prevalenceFull size tableExtended Data Table 2 Genomic sequence identities of bat SL-CoVs with SARS-CoVsFull size tableExtended Data Table 3 Genomic annotation and comparison of bat SL-CoV Rs3367 with human/civet SARS-CoVs and other bat SL-CoVsFull size tableExtended Data Table 4 Genomic annotation and comparison of bat SL-CoV RsSHC014 with human/civet SARS-CoVs and other bat SL-CoVsFull size tableExtended Data Table 5 Cell lines used for virus isolation and susceptibility testsFull size tablePowerPoint slides
PowerPoint slide for Fig. 1
PowerPoint slide for Fig. 2
PowerPoint slide for Fig. 3
PowerPoint slide for Fig. 4
Rights and permissions
Reprints and Permissions
About this article
Cite this article
Ge, X., Li, J., Yang, X. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013). https://doi.org/10.1038/nature12711
Download citation- Received16 May 2013
- Accepted18 September 2013
- Published30 October 2013
- Issue Date28 November 2013
- DOIhttps://doi.org/10.1038/nature12711
Anyone you share the following link with will be able to read this content:
Get shareable linkSubjectsFurther reading- Positive Selection of a Serine Residue in Bat IRF3 Confers Enhanced Antiviral Protection
- Arinjay Banerjee
- , Xi Zhang
- […]
- Karen Mossman
iScience (2020) - The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade
- B. Coutard
- , C. Valle
- […]
- E. Decroly
Antiviral Research (2020) - COVID-19: a novel zoonotic disease caused by a coronavirus from China: what we know and what we don't
- John S Mackenzie
- & David W Smith
Microbiology Australia (2020) - Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: Comparison with SARS and MERS
- Mohsen Rokni
- , Vida Ghasemi
- & Zahra Tavakoli
Reviews in Medical Virology (2020) - Bat‐borne viruses in Africa: a critical review
- W. Markotter
- , J. Coertse
- […]
- M. Mortlock
Journal of Zoology (2020)
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
Download PDFEditorial Summary
A SARS-like virus in bats
Peter Daszak and colleagues identify two novel coronaviruses from Chinese horseshoe bats that are closely related to severe acute… show more- Sections
- Figures
- References
- Abstract
- Main
- Methods Summary
- Online Methods
- Accession codes
- References
- Acknowledgements
- Author information
- Ethics declarations
- Extended data figures and tables
- PowerPoint slides
- Rights and permissions
- About this article
- Further reading
- Comments
-
The rest of the paper. Link above. I am claiming fair use due to the pertinence under the current laws of the United States. This is not a commercial use.- Letter
- Published: 30 October 2013
- Xing-Yi Ge,
- Jia-Lu Li,
- Xing-Lou Yang,
- Aleksei A. Chmura,
- Guangjian Zhu,
- Jonathan H. Epstein,
- Jonna K. Mazet,
- Ben Hu,
- Wei Zhang,
- Cheng Peng,
- Yu-Ji Zhang,
- Chu-Ming Luo,
- Bing Tan,
- Ning Wang,
- Yan Zhu,
- Gary Crameri,
- Shu-Yi Zhang,
- Lin-Fa Wang,
- Peter Daszak &
- Zheng-Li Shi
Nature volume 503, pages535–538(2013)Cite this article- 82k Accesses
- 327 Citations
- 1349 Altmetric
- Metricsdetails
The 2002–3 pandemic caused by severe acute respiratory syndrome coronavirus (SARS-CoV) was one of the most significant public health events in recent history1. An ongoing outbreak of Middle East respiratory syndrome coronavirus2 suggests that this group of viruses remains a key threat and that their distribution is wider than previously recognized. Although bats have been suggested to be the natural reservoirs of both viruses3,4,5, attempts to isolate the progenitor virus of SARS-CoV from bats have been unsuccessful. Diverse SARS-like coronaviruses (SL-CoVs) have now been reported from bats in China, Europe and Africa5,6,7,8, but none is considered a direct progenitor of SARS-CoV because of their phylogenetic disparity from this virus and the inability of their spike proteins to use the SARS-CoV cellular receptor molecule, the human angiotensin converting enzyme II (ACE2)9,10.
Here we report whole-genome sequences of two novel bat coronaviruses from Chinese horseshoe bats (family: Rhinolophidae) in Yunnan, China: RsSHC014 and Rs3367. These viruses are far more closely related to SARS-CoV than any previously identified bat coronaviruses, particularly in the receptor binding domain of the spike protein. Most importantly, we report the first recorded isolation of a live SL-CoV (bat SL-CoV-WIV1) from bat faecal samples in Vero E6 cells, which has typical coronavirus morphology, 99.9% sequence identity to Rs3367 and uses ACE2 from humans, civets and Chinese horseshoe bats for cell entry.
Preliminary in vitro testing indicates that WIV1 also has a broad species tropism. Our results provide the strongest evidence to date that Chinese horseshoe bats are natural reservoirs of SARS-CoV, and that intermediate hosts may not be necessary for direct human infection by some bat SL-CoVs.
They also highlight the importance of pathogen-discovery programs targeting high-risk wildlife groups in emerging disease hotspots as a strategy for pandemic preparedness.
Main
The 2002–3 pandemic of SARS1 and the ongoing emergence of the Middle East respiratory syndrome coronavirus (MERS-CoV)2 demonstrate that CoVs are a significant public health threat. SARS-CoV was shown to use the human ACE2 molecule as its entry receptor, and this is considered a hallmark of its cross-species transmissibility11. The receptor binding domain (RBD) located in the amino-terminal region (amino acids 318–510) of the SARS-CoV spike (S) protein is directly involved in binding to ACE2 (ref. 12). However, despite phylogenetic evidence that SARS-CoV evolved from bat SL-CoVs, all previously identified SL-CoVs have major sequence differences from SARS-CoV in the RBD of their S proteins, including one or two deletions6,9. Replacing the RBD of one SL-CoV S protein with SARS-CoV S conferred the ability to use human ACE2 and replicate efficiently in mice9,13. However, to date, no SL-CoVs have been isolated from bats, and no wild-type SL-CoV of bat origin has been shown to use ACE2.
We conducted a 12-month longitudinal survey (April 2011–September 2012) of SL-CoVs in a colony of Rhinolophus sinicus at a single location in Kunming, Yunnan Province, China (Extended Data Table 1). A total of 117 anal swabs or faecal samples were collected from individual bats using a previously published method5,14. A one-step reverse transcription (RT)-nested PCR was conducted to amplify the RNA-dependent RNA polymerase (RdRP) motifs A and C, which are conserved among alphacoronaviruses and betacoronaviruses15.
Twenty-seven of the 117 samples (23%) were classed as positive by PCR and subsequently confirmed by sequencing. The species origin of all positive samples was confirmed to be R. sinicus by cytochrome b sequence analysis, as described previously16. A higher prevalence was observed in samples collected in October (30% in 2011 and 48.7% in 2012) than those in April (7.1% in 2011) or May (7.4% in 2012) (Extended Data Table 1). Analysis of the S protein RBD sequences indicated the presence of seven different strains of SL-CoVs (Fig. 1a and Extended Data Figs 1 and 2). In addition to RBD sequences, which closely matched previously described SL-CoVs (Rs672, Rf1 and HKU3)5,8,17,18, two novel strains (designated SL-CoV RsSHC014 and Rs3367) were discovered. Their full-length genome sequences were determined, and both were found to be 29,787 base pairs in size (excluding the poly(A) tail). The overall nucleotide sequence identity of these two genomes with human SARS-CoV (Tor2 strain) is 95%, higher than that observed previously for bat SL-CoVs in China (88–92%)5,8,17,18 or Europe (76%)6 (Extended Data Table 2 and Extended Data Figs 3 and 4). Higher sequence identities were observed at the protein level between these new SL-CoVs and SARS-CoVs (Extended Data Tables 3 and 4). To understand the evolutionary origin of these two novel SL-CoV strains, we conducted recombination analysis with the Recombination Detection Program 4.0 package19 using available genome sequences of bat SL-CoV strains (Rf1, Rp3, Rs672, Rm1, HKU3 and BM48-31) and human and civet representative SARS-CoV strains (BJ01, SZ3, Tor2 and GZ02). Three breakpoints were detected with strong P values (<10−20) and supported by similarity plot and bootscan analysis (Extended Data Fig. 5a, b). Breakpoints were located at nucleotides 20,827, 26,553 and 28,685 in the Rs3367 (and RsSHC014) genome, and generated recombination fragments covering nucleotides 20,827–26,533 (5,727 nucleotides) (including partial open reading frame (ORF) 1b, full-length S, ORF3, E and partial M gene) and nucleotides 26,534–28,685 (2,133 nucleotides) (including partial ORF M, full-length ORF6, ORF7, ORF8 and partial N gene). Phylogenetic analysis using the major and minor parental regions suggested that Rs3367, or RsSHC014, is the descendent of a recombination of lineages that ultimately lead to SARS-CoV and SL-CoV Rs672 (Fig. 1b).
Figure 1: Phylogenetic tree based on amino acid sequences of the S RBD region and the two parental regions of bat SL-CoV Rs3367 or RsSHC014.
a, SARS-CoV S protein amino acid residues 310–520 were aligned with homologous regions of bat SL-CoVs using the ClustalW software. A maximum-likelihood phylogenetic tree was constructed using a Poisson model with bootstrap values determined by 1,000 replicates in the MEGA5 software package. The RBD sequences identified in this study are in bold and named by the sample numbers. The key amino acid residues involved in interacting with the human ACE2 molecule are indicated on the right of the tree. SARS-CoV GZ02, BJ01 and Tor2 were isolated from patients in the early, middle and late phase, respectively, of the SARS outbreak in 2003. SARS-CoV SZ3 was identified from Paguma larvata in 2003 collected in Guangdong, China. SL-CoV Rp3, Rs672 and HKU3-1 were identified from R. sinicus collected in China (respectively: Guangxi, 2004; Guizhou, 2006; Hong Kong, 2005). Rf1 and Rm1 were identified from R. ferrumequinum and R. macrotis, respectively, collected in Hubei, China, in 2004. Bat SARS-related CoV BM48-31 was identified from R. blasii collected in Bulgaria in 2008. Bat CoV HKU9-1 was identified from Rousettus leschenaultii collected in Guangdong, China in 2005/2006 and used as an outgroup. All sequences in bold and italics were identified in the current study. Filled triangles, circles and diamonds indicate samples with co-infection by two different SL-CoVs. ‘–’ indicates the amino acid deletion. b, Phylogenetic origins of the two parental regions of Rs3367 or RsSHC014. Maximum likelihood phylogenetic trees were constructed from alignments of two fragments covering nucleotides 20,827–26,533 (5,727 nucleotides) and 26,534 –28,685 (2,133 nucleotides) of the Rs3367 genome, respectively. For display purposes, the trees were midpoint rooted. The taxa were annotated according to strain names: SARS-CoV, SARS coronavirus; SARS-like CoV, bat SARS-like coronavirus. The two novel SL-CoVs, Rs3367 and RsSHC014, are in bold and italics.
PowerPoint slide
Full size image
The most notable sequence differences between these two new SL-CoVs and previously identified SL-CoVs is in the RBD regions of their S proteins. First, they have higher amino acid sequence identity to SARS-CoV (85% and 96% for RsSHC014 and Rs3367, respectively). Second, there are no deletions and they have perfect sequence alignment with the SARS-CoV RBD region (Extended Data Figs 1 and 2). Structural and mutagenesis studies have previously identified five key residues (amino acids 442, 472, 479, 487 and 491) in the RBD of the SARS-CoV S protein that have a pivotal role in receptor binding20,21. Although all five residues in the RsSHC014 S protein were found to be different from those of SARS-CoV, two of the five residues in the Rs3367 RBD were conserved (Fig. 1 and Extended Data Fig. 1).
Despite the rapid accumulation of bat CoV sequences in the last decade, there has been no report of successful virus isolation6,22,23. We attempted isolation from SL-CoV PCR-positive samples. Using an optimized protocol and Vero E6 cells, we obtained one isolate which caused cytopathic effect during the second blind passage. Purified virions displayed typical coronavirus morphology under electron microscopy (Fig. 2). Sequence analysis using a sequence-independent amplification method14 to avoid PCR-introduced contamination indicated that the isolate was almost identical to Rs3367, with 99.9% nucleotide genome sequence identity and 100% amino acid sequence identity for the S1 region. The new isolate was named SL-CoV-WIV1.
Figure 2: Electron micrograph of purified virions.
Virions from a 10-ml culture were collected, fixed and concentrated/purified by sucrose gradient centrifugation. The pelleted viral particles were suspended in 100 μl PBS, stained with 2% phosphotungstic acid (pH 7.0) and examined directly using a Tecnai transmission electron microscope (FEI) at 200 kV.
PowerPoint slide
Full size image
To determine whether WIV1 can use ACE2 as a cellular entry receptor, we conducted virus infectivity studies using HeLa cells expressing or not expressing ACE2 from humans, civets or Chinese horseshoe bats. We found that WIV1 is able to use ACE2 of different origins as an entry receptor and replicated efficiently in the ACE2-expressing cells (Fig. 3). This is, to our knowledge, the first identification of a wild-type bat SL-CoV capable of using ACE2 as an entry receptor.
Figure 3: Analysis of receptor usage of SL-CoV-WIV1 determined by immunofluorescence assay and real-time PCR.
Determination of virus infectivity in HeLa cells with and without the expression of ACE2. b, bat; c, civet; h, human. ACE2 expression was detected with goat anti-humanACE2 antibody followed by fluorescein isothiocyanate (FITC)-conjugated donkey anti-goat IgG. Virus replication was detected with rabbit antibody against the SL-CoV Rp3 nucleocapsid protein followed by cyanine 3 (Cy3)-conjugated mouse anti-rabbit IgG. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). The columns (from left to right) show staining of nuclei (blue), ACE2 expression (green), virus replication (red), merged triple-stained images and real-time PCR results, respectively. (n = 3); error bars represent standard deviation.
PowerPoint slide
Full size image
To assess its cross-species transmission potential, we conducted infectivity assays in cell lines from a range of species. Our results (Fig. 4 and Extended Data Table 5) indicate that bat SL-CoV-WIV1 can grow in human alveolar basal epithelial (A549), pig kidney 15 (PK-15) and Rhinolophus sinicus kidney (RSKT) cell lines, but not in human cervix (HeLa), Syrian golden hamster kidney (BHK21), Myotis davidii kidney (BK), Myotis chinensis kidney (MCKT), Rousettus leschenaulti kidney (RLK) or Pteropus alecto kidney (PaKi) cell lines. Real-time RT–PCR indicated that WIV1 replicated much less efficiently in A549, PK-15 and RSKT cells than in Vero E6 cells (Fig. 4).
Figure 4: Analysis of host range of SL-CoV-WIV1 determined by immunofluorescence assay and real-time PCR.
Virus infection in A549, RSKT, Vero E6 and PK-15 cells. Virus replication was detected as described for Fig. 3. The columns (from left to right) show staining of nuclei (blue), virus replication (red), merged double-stained images and real-time PCR results, respectively. n = 3; error bars represent s.d.
PowerPoint slide
Full size image
To assess the cross-neutralization activity of human SARS-CoV sera against WIV1, we conducted serum-neutralization assays using nine convalescent sera from SARS patients collected in 2003. The results showed that seven of these were able to completely neutralize 100 tissue culture infectious dose 50 (TCID50) WIV1 at dilutions of 1:10 to 1:40, further confirming the close relationship between WIV1 and SARS-CoV.
Our findings have important implications for public health. First, they provide the clearest evidence yet that SARS-CoV originated in bats. Our previous work provided phylogenetic evidence of this5, but the lack of an isolate or evidence that bat SL-CoVs can naturally infect human cells, until now, had cast doubt on this hypothesis. Second, the lack of capacity of SL-CoVs to use of ACE2 receptors has previously been considered as the key barrier for their direct spillover into humans, supporting the suggestion that civets were intermediate hosts for SARS-CoV adaptation to human transmission during the SARS outbreak24. However, the ability of SL-CoV-WIV1 to use human ACE2 argues against the necessity of this step for SL-CoV-WIV1 and suggests that direct bat-to-human infection is a plausible scenario for some bat SL-CoVs. This has implications for public health control measures in the face of potential spillover of a diverse and growing pool of recently discovered SARS-like CoVs with a wide geographic distribution.
Our findings suggest that the diversity of bat CoVs is substantially higher than that previously reported. In this study we were able to demonstrate the circulation of at least seven different strains of SL-CoVs within a single colony of R. sinicus during a 12-month period. The high genetic diversity of SL-CoVs within this colony was mirrored by high phenotypic diversity in the differential use of ACE2 by different strains. It would therefore not be surprising if further surveillance reveals a broad diversity of bat SL-CoVs that are able to use ACE2, some of which may have even closer homology to SARS-CoV than SL-CoV-WIV1. Our results—in addition to the recent demonstration of MERS-CoV in a Saudi Arabian bat25, and of bat CoVs closely related to MERS-CoV in China, Africa, Europe and North America3,26,27—suggest that bat coronaviruses remain a substantial global threat to public health.
Finally, this study demonstrates the public health importance of pathogen discovery programs targeting wildlife that aim to identify the ‘known unknowns’—previously unknown viral strains closely related to known pathogens. These programs, focused on specific high-risk wildlife groups and hotspots of disease emergence, may be a critical part of future global strategies to predict, prepare for, and prevent pandemic emergence28.
Methods Summary
Throat and faecal swabs or fresh faecal samples were collected in viral transport medium as described previously14. All PCR was conducted with the One-Step RT–PCR kit (Invitrogen). Primers targeting the highly conserved regions of the RdRP gene were used for detection of all alphacoronaviruses and betacoronaviruses as described previously15. Degenerate primers were designed on the basis of all available genomic sequences of SARS-CoVs and SL-CoVs and used for amplification of the RBD sequences of S genes or full-length genomic sequences. Degenerate primers were used for amplification of the bat ACE2 gene as described previously29. PCR products were gel purified and cloned into pGEM-T Easy Vector (Promega). At least four independent clones were sequenced to obtain a consensus sequence. PCR-positive faecal samples (in 200 μl buffer) were gradient centrifuged at 3,000–12,000g and supernatant diluted at 1:10 in DMEM before being added to Vero E6 cells. After incubation at 37 ?C for 1 h, inocula were removed and replaced with fresh DMEM with 2% FCS. Cells were incubated at 37 ?C and checked daily for cytopathic effect. Cell lines from different origins were grown on coverslips in 24-well plates and inoculated with the novel SL-CoV at a multiplicity of infection of 10. Virus replication was detected at 24 h after infection using rabbit antibodies against the SL-CoV Rp3 nucleocapsid protein followed by Cy3-conjugated goat anti-rabbit IgG.
Online Methods
Sampling
Bats were trapped in their natural habitat as described previously5. Throat and faecal swab samples were collected in viral transport medium (VTM) composed of Hank’s balanced salt solution, pH 7.4, containing BSA (1%), amphotericin (15 μg ml−1), penicillin G (100 U ml−1) and streptomycin (50 μg ml−1). To collect fresh faecal samples, clean plastic sheets measuring 2.0 by 2.0 m were placed under known bat roosting sites at about 18:00 h each evening. Relatively fresh faecal samples were collected from sheets at approximately 05:30–06:00 the next morning and placed in VTM. Samples were transported to the laboratory and stored at −80 ?C until use. All animals trapped for this study were released back to their habitat after sample collection. All sampling processes were performed by veterinarians with approval from Animal Ethics Committee of the Wuhan Institute of Virology (WIVH05210201) and EcoHealth Alliance under an inter-institutional agreement with University of California, Davis (UC Davis protocol no. 16048).
RNA extraction, PCR and sequencing
RNA was extracted from 140 μl of swab or faecal samples with a Viral RNA Mini Kit (Qiagen) following the manufacturer’s instructions. RNA was eluted in 60 μl RNAse-free buffer (buffer AVE, Qiagen), then aliquoted and stored at −80 ?C. One-step RT–PCR (Invitrogen) was used to detect coronavirus sequences as described previously15. First round PCR was conducted in a 25-μl reaction mix containing 12.5 μl PCR 2? reaction mix buffer, 10 pmol of each primer, 2.5 mM MgSO4, 20 U RNase inhibitor, 1 μl SuperScript III/ Platinum Taq Enzyme Mix and 5 μl RNA. Amplification of the RdRP-gene fragment was performed as follows: 50 ?C for 30 min, 94 ?C for 2 min, followed by 40 cycles consisting of 94 ?C for 15 s, 62 ?C for 15 s, 68 ?C for 40 s, and a final extension of 68 ?C for 5 min. Second round PCR was conducted in a 25-μl reaction mix containing 2.5 μl PCR reaction buffer, 5 pmol of each primer, 50 mM MgCl2, 0.5 mM dNTP, 0.1 μl Platinum Taq Enzyme (Invitrogen) and 1 μl first round PCR product. The amplification of RdRP-gene fragment was performed as follows: 94 ?C for 5 min followed by 35 cycles consisting of 94 ?C for 30 s, 52 ?C for 30 s, 72 ?C for 40 s, and a final extension of 72 ?C for 5 min.
To amplify the RBD region, one-step RT–PCR was performed with primers designed based on available SARS-CoV or bat SL-CoVs (first round PCR primers; F, forward; R, reverse: CoVS931F-5′-VWGADGTTGTKAGRTTYCCT-3′ and CoVS1909R-5′-TAARACAVCCWGCYTGWGT-3′; second PCR primers: CoVS951F-5′-TGTKAGRTTYCCTAAYATTAC-3′ and CoVS1805R-5′-ACATCYTGATANARAACAGC-3′). First-round PCR was conducted in a 25-μl reaction mix as described above except primers specific for the S gene were used. The amplification of the RBD region of the S gene was performed as follows: 50 ?C for 30 min, 94 ?C for 2 min, followed by 35 cycles consisting of 94 ?C for 15 s, 43 ?C for 15 s, 68 ?C for 90 s, and a final extension of 68 ?C for 5 min. Second-round PCR was conducted in a 25-μl reaction mix containing 2.5 μl PCR reaction buffer, 5 pmol of each primer, 50 mM MgCl2, 0.5 mM dNTP, 0.1 μl Platinum Taq Enzyme (Invitrogen) and 1 μl first round PCR product. Amplification was performed as follows: 94 ?C for 5 min followed by 40 cycles consisting of 94 ?C for 30 s, 41 ?C for 30 s, 72 ?C for 60 s, and a final extension of 72 ?C for 5 min.
PCR products were gel purified and cloned into pGEM-T Easy Vector (Promega). At least four independent clones were sequenced to obtain a consensus sequence for each of the amplified regions.
Sequencing full-length genomes
Degenerate coronavirus primers were designed based on all available SARS-CoV and bat SL-CoV sequences in GenBank and specific primers were designed from genome sequences generated from previous rounds of sequencing in this study (primer sequences will be provided upon request). All PCRs were conducted using the One-Step RT–PCR kit (Invitrogen). The 5′ and 3′ genomic ends were determined using the 5′ or 3′ RACE kit (Roche), respectively. PCR products were gel purified and sequenced directly or following cloning into pGEM-T Easy Vector (Promega). At least four independent clones were sequenced to obtain a consensus sequence for each of the amplified regions and each region was sequenced at least twice.
Sequence analysis and databank accession numbers
Routine sequence management and analysis was carried out using DNAStar or Geneious. Sequence alignment and editing was conducted using ClustalW, BioEdit or GeneDoc. Maximum Likelihood phylogenetic trees based on the protein sequences were constructed using a Poisson model with bootstrap values determined by 1,000 replicates in the MEGA5 software package.
Sequences obtained in this study have been deposited in GenBank as follows (accession numbers given in parenthesis): full-length genome sequence of SL-CoV RsSHC014 and Rs3367 (KC881005, KC881006); full-length sequence of WIV1 S (KC881007); RBD (KC880984-KC881003); ACE2 (KC8810040). SARS-CoV sequences used in this study: human SARS-CoV strains Tor2 (AY274119), BJ01 (AY278488), GZ02 (AY390556) and civet SARS-CoV strain SZ3 (AY304486). Bat coronavirus sequences used in this study: Rs672 (FJ588686), Rp3 (DQ071615), Rf1 (DQ412042), Rm1 (DQ412043), HKU3-1 (DQ022305), BM48-31 (NC_014470), HKU9-1 (NC_009021), HKU4 (NC_009019), HKU5 (NC_009020), HKU8 (DQ249228), HKU2 (EF203067), BtCoV512 (NC_009657), 1A (NC_010437). Other coronavirus sequences used in this study: HCoV-229E (AF304460), HCoV-OC43 (AY391777), HCoV-NL63 (AY567487), HKU1 (NC_006577), EMC (JX869059), FIPV (NC_002306), PRCV (DQ811787), BWCoV (NC_010646), MHV (AY700211), IBV (AY851295).
Amplification, cloning and expression of the bat ACE2 gene
Construction of expression clones for human and civet ACE2 in pcDNA3.1 has been described previously29. Bat ACE2 was amplified from a R. sinicus (sample no. 3357). In brief, total RNA was extracted from bat rectal tissue using the RNeasy Mini Kit (Qiagen). First-strand complementary DNA was synthesized from total RNA by reverse transcription with random hexamers. Full-length bat ACE2 fragments were amplified using forward primer bAF2 and reverse primer bAR2 (ref. 29). The ACE2 gene was cloned into pCDNA3.1 with KpnI and XhoI, and verified by sequencing. Purified ACE2 plasmids were transfected to HeLa cells. After 24 h, lysates of HeLa cells expressing human, civet, or bat ACE2 were confirmed by western blot or immunofluorescence assay.
Western blot analysis
Lysates of cells or filtered supernatants containing pseudoviruses were separated by SDS–PAGE, followed by transfer to a nitrocellulose membrane (Millipore). For detection of S protein, the membrane was incubated with rabbit anti-Rp3 S fragment (amino acids 561–666) polyantibodies (1:200), and the bound antibodies were detected by alkaline phosphatase (AP)-conjugated goat anti-rabbit IgG (1:1,000). For detection of HIV-1 p24 in supernatants, monoclonal antibody against HIV p24 (p24 MAb) was used as the primary antibody at a dilution of 1:1,000, followed by incubation with AP-conjugated goat anti-mouse IgG at the same dilution. To detect the expression of ACE2 in HeLa cells, goat antibody against the human ACE2 ectodomain (1:500) was used as the first antibody, followed by incubation with horseradish peroxidase-conjugated donkey anti-goat IgG (1:1,000).
Virus isolation
Vero E6 cell monolayers were maintained in DMEM supplemented with 10% FCS. PCR-positive samples (in 200 μl buffer) were gradient centrifuged at 3,000–12,000g, and supernatant were diluted 1:10 in DMEM before being added to Vero E6 cells. After incubation at 37 ?C for 1 h, inocula were removed and replaced with fresh DMEM with 2% FCS. Cells were incubated at 37 ?C for 3 days and checked daily for cytopathic effect. Double-dose triple antibiotics penicillin/streptomycin/amphotericin (Gibco) were included in all tissue culture media (penicillin 200 IU ml−1, streptomycin 0.2 mg ml−1, amphotericin 0.5 μg ml−1). Three blind passages were carried out for each sample. After each passage, both the culture supernatant and cell pellet were examined for presence of virus by RT–PCR using primers targeting the RdRP or S gene. Virions in supernatant (10 ml) were collected and fixed using 0.1% formaldehyde for 4 h, then concentrated by ultracentrifugation through a 20% sucrose cushion (5 ml) at 80,000g for 90 min using a Ty90 rotor (Beckman). The pelleted viral particles were suspended in 100 μl PBS, stained with 2% phosphotungstic acid (pH 7.0) and examined using a Tecnai transmission electron microscope (FEI) at 200 kV.
Virus infectivity detected by immunofluorescence assay
Cell lines used for this study and their culture conditions are summarized in Extended Data Table 5. Virus titre was determined in Vero E6 cells by cytopathic effect (CPE) counts. Cell lines from different origins and HeLa cells expressing ACE2 from human, civet or Chinese horseshoe bat were grown on coverslips in 24-well plates (Corning) incubated with bat SL-CoV-WIV1 at a multiplicity of infection = 10 for 1 h. The inoculum was removed and washed twice with PBS and supplemented with medium. HeLa cells without ACE2 expression and Vero E6 cells were used as negative and positive controls, respectively. At 24 h after infection, cells were washed with PBS and fixed with 4% formaldehyde in PBS (pH 7.4) for 20 min at 4 ?C. ACE2 expression was detected using goat anti-human ACE2 immunoglobulin (R&D Systems) followed by FITC-labelled donkey anti-goat immunoglobulin (PTGLab). Virus replication was detected using rabbit antibody against the SL-CoV Rp3 nucleocapsid protein followed by Cy3-conjugated mouse anti-rabbit IgG. Nuclei were stained with DAPI. Staining patterns were examined using a FV1200 confocal microscope (Olympus).
Virus infectivity detected by real-time RT–PCR
Vero E6, A549, PK15, RSKT and HeLa cells with or without expression of ACE2 of different origins were inoculated with 0.1 TCID50 WIV-1 and incubated for 1 h at 37 ?C. After removing the inoculum, the cells were cultured with medium containing 1% FBS. Supernatants were collected at 0, 12, 24 and 48 h. RNA from 140 μl of each supernatant was extracted with the Viral RNA Mini Kit (Qiagen) following manufacturer’s instructions and eluted in 60 μl buffer AVE (Qiagen). RNA was quantified on the ABI StepOne system, with the TaqMan AgPath-ID One-Step RT–PCR Kit (Applied Biosystems) in a 25 μl reaction mix containing 4 μl RNA, 1 ? RT–PCR enzyme mix, 1 ? RT–PCR buffer, 40 pmol forward primer (5′-GTGGTGGTGACGGCAAAATG-3′), 40 pmol reverse primer (5′-AAGTGAAGCTTCTGGGCCAG-3′) and 12 pmol probe (5′-FAM-AAAGAGCTCAGCCCCAGATG-BHQ1-3′). Amplification parameters were 10 min at 50 ?C, 10 min at 95 ?C and 50 cycles of 15 s at 95 ?C and 20 s at 60 ?C. RNA dilutions from purified WIV-1 stock were used as a standard.
Serum neutralization test
SARS patient sera were inactivated at 56 ?C for 30 min and then used for virus neutralization testing. Sera were diluted starting with 1:10 and then serially twofold diluted in 96-well cell plates to 1:40. Each 100 μl serum dilution was mixed with 100 μl viral supernatant containing 100 TCID50of WIV1 and incubated at 37 ?C for 1 h. The mixture was added in triplicate wells of 96-well cell plates with plated monolayers of Vero E6 cells and further incubated at 37 ?C for 2 days. Serum from a healthy blood donor was used as a negative control in each experiment. CPE was observed using an inverted microscope 2 days after inoculation. The neutralizing antibody titre was read as the highest dilution of serum which completely suppressed CPE in infected wells. The neutralization test was repeated twice.
Recombination analysis
Full-length genomic sequences of SL-CoV Rs3367 or RsSHC014 were aligned with those of selected SARS-CoVs and bat SL-CoVs using Clustal X. The aligned sequences were preliminarily scanned for recombination events using Recombination Detection Program (RDP) 4.0 (ref. 19). The potential recombination events suggested by RDP owing to their strong P values (<10–20) were investigated further by similarity plot and bootscan analyses implemented in Simplot 3.5.1. Phylogenetic origin of the major and minor parental regions of Rs3367 or RsSHC014 were constructed from the concatenated sequences of the essential ORFs of the major and minor parental regions of selected SARS-CoV and SL-CoVs. Two genome regions between three estimated breakpoints (20,827–26,553 and 26,554–28,685) were aligned independently using ClustalX and generated two alignments of 5,727 base pairs and 2,133 base pairs. The two alignments were used to construct maximum likelihood trees to better infer the fragment parents. All nucleotide numberings in this study are based on Rs3367 genome position.
Accession codes
Accessions
GenBank/EMBL/DDBJData deposits
Sequences of three bat SL-CoV genomes, bat SL-CoV RBD and R. sinicus ACE2 genes have been deposited in GenBank under accession numbers KC881005–KC881007 (genomes from SL-CoV RsSHC014, Rs3367 and W1V1, respectively), KC880984–KC881003 (bat SL-CoV RBD genes) and KC881004 (R. sinicus ACE2), respectively.
References- 1
Ksiazek, T. G. et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966 (2003) - 2
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. & Fouchier, R. A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820 (2012) - 3
Anthony, S. J. et al. Coronaviruses in bats from Mexico. J. Gen. Virol. 94, 1028–1038 (2013) - 4
Raj, V. S. et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495, 251–254 (2013) - 5
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005) - 6
Drexler, J. F. et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 84, 11336–11349 (2010) - 7
Tong, S. et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15, 482–485 (2009) - 8
Lau, S. K. P. et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl Acad. Sci. USA 102, 14040–14045 (2005) - 9
Ren, W. et al. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 82, 1899–1907 (2008) - 10
Hon, C. C. et al. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 82, 1819–1826 (2008) - 11
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003) - 12
Wong, S. K., Li, W., Moore, M. J., Choe, H. & Farzan, M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 279, 3197–3201 (2004) - 13
Becker, M. M. et al. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl Acad. Sci. USA 105, 19944–19949 (2008) - 14
Li, Y. et al. Host range, prevalence, and genetic diversity of adenoviruses in bats. J. Virol. 84, 3889–3897 (2010) - 15
De Souza Luna, L. K. et al. Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J. Clin. Microbiol. 45, 1049–1052 (2007) - 16
Cui, J. et al. Evolutionary relationships between bat coronaviruses and their hosts. Emerg. Infect. Dis. 13, 1526–1532 (2007) - 17
Yuan, J. et al. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 91, 1058–1062 (2010) - 18
Ren, W. et al. Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J. Gen. Virol. 87, 3355–3359 (2006) - 19
Martin, D. P. et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26, 2462–2463 (2010) - 20
Wu, K., Peng, G., Wilken, M., Geraghty, R. J. & Li, F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 287, 8904–8911 (2012) - 21
Li, W. et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 24, 1634–1643 (2005) - 22
Lau, S. K. et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 84, 2808–2819 (2010) - 23
Lau, S. K. et al. Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J. Virol. 84, 11385–11394 (2010) - 24
Song, H. D. et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl Acad. Sci. USA 102, 2430–2435 (2005) - 25
Memish, Z. A. et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 19, 11 (2013) - 26
Chan, J. F. et al. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 65, 477–489 (2012) - 27
Ithete, N. L. et al. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 19, 1697–1699 (2013) - 28
Morse, S. S. et al. Prediction and prevention of the next pandemic zoonosis. Lancet 380, 1956–1965 (2012) - 29
Hou, Y. et al. Angiotensin-converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS-CoV entry. Arch. Virol. 155, 1563–1569 (2010)
Download references
Acknowledgements
We acknowledge financial support from the State Key Program for Basic Research (2011CB504701 and 2010CB530100), National Natural Science Foundation of China (81290341 and 31321001), Scientific and technological basis special project (2013FY113500), CSIRO OCE Science Leaders Award, National Institute of Allergy and Infectious Diseases (NIAID) award number R01AI079231, a National Institutes of Health (NIH)/National Science Foundation (NSF) ‘Ecology and Evolution of Infectious Diseases’ award from the NIH Fogarty International Center (R01TW005869), an award from the NIH Fogarty International Center supported by International Influenza Funds from the Office of the Secretary of the Department of Health and Human Services (R56TW009502), and United States Agency for International Development (USAID) Emerging Pandemic Threats PREDICT. The contents are the responsibility of the authors and do not necessarily reflect the views of NIAID, NIH, NSF, USAID or the United States Government. We thank X. Che from Zhujiang Hospital, Southern Medical University, for providing human SARS patient sera.
Author information
Author notes- Xing-Yi Ge, Jia-Lu Li and Xing-Lou Yang: These authors contributed equally to this work.
- Center for Emerging Infectious Diseases, State Key Laboratory of Virology, Wuhan Institute of Virology of the Chinese Academy of Sciences, Wuhan, 430071, China
- Xing-Yi Ge
- , Jia-Lu Li
- , Xing-Lou Yang
- , Ben Hu
- , Wei Zhang
- , Cheng Peng
- , Yu-Ji Zhang
- , Chu-Ming Luo
- , Bing Tan
- , Ning Wang
- , Yan Zhu
- & Zheng-Li Shi
- EcoHealth Alliance, New York, 10001, New York, USA
- Aleksei A. Chmura
- , Guangjian Zhu
- , Jonathan H. Epstein
- & Peter Daszak
- One Health Institute, School of Veterinary Medicine, University of California, Davis, 95616, California, USA
- Jonna K. Mazet
- CSIRO Australian Animal Health Laboratory, Geelong, 3220, Victoria, Australia
- Gary Crameri
- & Lin-Fa Wang
- College of Life Sciences, East China Normal University, Shanghai 200062, China
- Shu-Yi Zhang
- Emerging Infectious Diseases Program, Duke-NUS Graduate Medical School, Singapore 169857
- Lin-Fa Wang
Z.-L.S. and P.D. designed and coordinated the study. X.-Y.G., J.-L. L. and X.-L.Y. conducted majority of experiments and contributed equally to the study. A.A.C., B.H., W.Z., C.P., Y.-J.Z., C.-M.L., B.T., N.W. and Y.Z. conducted parts of the experiments and analyses. J.H.E., J.K.M. and S.-Y.Z. coordinated the field study. X.-Y.G., J.-L.L., X.-L.Y., B.T. and G.-J.Z. collected the samples. G.C. and L.-F.W. designed and supervised part of the experiments. All authors contributed to the interpretations and conclusions presented. Z.-L.S. and X-Y.G. wrote the manuscript with significant contributions from P.D. and L-F.W. and input from all authors.
Corresponding authors
Correspondence to Peter Daszak or Zheng-Li Shi.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Sequence alignment of CoV S protein RBD.
SARS-CoV S protein (amino acids 310–520) is aligned with homologous regions of bat SL-CoVs using ClustalW. The newly discovered bat SL-CoVs are indicated with a bold vertical line on the left. The key amino acid residues involved in the interaction with human ACE2 are numbered on the top of the aligned sequences.
Extended Data Figure 2 Alignment of CoV S protein S1 sequences.
Alignment of S1 sequences (amino acids 1–660) of the two novel bat SL-CoV S proteins with those of previously reported bat SL-CoVs and human and civet SARS-CoVs. The newly discovered bat SL-CoVs are boxed in red. SARS-CoV GZ02, BJ01 and Tor2 were isolated from patients in the early, middle and late phase, respectively, of the SARS outbreak in 2003. SARS-CoV SZ3 was identified from P. larvata in 2003 collected in Guangdong, China. SL-CoV Rp3, Rs 672 and HKU3-1 were identified from R. sinicus collected in Guangxi, Guizhou and Hong Kong, China, respectively. Rf1 and Rm1 were identified from R. ferrumequinum and R. macrotis, respectively, collected in Hubei Province, China. Bat SARS-related CoV BM48-31 was identified from R. blasii collected in Bulgaria.
Extended Data Figure 3 Complete RdRP sequence phylogeny.
Phylogenetic tree of bat SL-CoVs and SARS-CoVs on the basis of complete RdRP sequences (2,796 nucleotides). Bat SL-CoVs RsSHC014 and Rs3367 are highlighted by filled circles. Three established coronaivirus genera, Alphacoronavirus, Betacoronavirus and Gammacoronavirus are marked as α, β and γ, respectively. Four CoV groups in the genus Betacoronavirus are indicated as A, B, C and D, respectively. MHV, murine hepatitis virus; PHEV, porcine haemagglutinating encephalomyelitis virus; PRCV, porcine respiratory coronavirus; FIPV, feline infectious peritonitis virus; IBV, infectious bronchitis coronavirus; BW, beluga whale coronavirus.
Extended Data Figure 4 Sequence phylogeny of the complete S protein of SL-CoVs and SARS-CoV.
Phylogenetic tree of bat SL-CoVs and SARS-CoVs on the basis of complete S protein sequences (1,256 amino acids). Bat SL-CoVs RsSHC014 and Rs3367 are highlighted by filled circles. Bat CoV HKU9 was used as an outgroup.
Extended Data Figure 5 Detection of potential recombination events.
a, b, Similarity plot (a) and bootscan analysis (b) detected three recombination breakpoints in the bat SL-CoV Rs3367 or SHC014 genome. The three breakpoints were located at the ORF1b (nt 20,827), M (nucleotides 26,553) and N (nucleotides 28,685) genes, respectively. Both analyses were performed with an F84 distance model, a window size of 1,500 base pairs and a step size of 300 base pairs.
Extended Data Table 1 Summary of sampling detail and CoV prevalenceFull size tableExtended Data Table 2 Genomic sequence identities of bat SL-CoVs with SARS-CoVsFull size tableExtended Data Table 3 Genomic annotation and comparison of bat SL-CoV Rs3367 with human/civet SARS-CoVs and other bat SL-CoVsFull size tableExtended Data Table 4 Genomic annotation and comparison of bat SL-CoV RsSHC014 with human/civet SARS-CoVs and other bat SL-CoVsFull size tableExtended Data Table 5 Cell lines used for virus isolation and susceptibility testsFull size tablePowerPoint slides
PowerPoint slide for Fig. 1
PowerPoint slide for Fig. 2
PowerPoint slide for Fig. 3
PowerPoint slide for Fig. 4
Rights and permissions
Reprints and Permissions
About this article
Cite this article
Ge, X., Li, J., Yang, X. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013). https://doi.org/10.1038/nature12711
Download citation- Received16 May 2013
- Accepted18 September 2013
- Published30 October 2013
- Issue Date28 November 2013
- DOIhttps://doi.org/10.1038/nature12711
Anyone you share the following link with will be able to read this content:
Get shareable linkSubjectsFurther reading- Positive Selection of a Serine Residue in Bat IRF3 Confers Enhanced Antiviral Protection
- Arinjay Banerjee
- , Xi Zhang
- […]
- Karen Mossman
iScience (2020) - The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade
- B. Coutard
- , C. Valle
- […]
- E. Decroly
Antiviral Research (2020) - COVID-19: a novel zoonotic disease caused by a coronavirus from China: what we know and what we don't
- John S Mackenzie
- & David W Smith
Microbiology Australia (2020) - Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: Comparison with SARS and MERS
- Mohsen Rokni
- , Vida Ghasemi
- & Zahra Tavakoli
Reviews in Medical Virology (2020) - Bat‐borne viruses in Africa: a critical review
- W. Markotter
- , J. Coertse
- […]
- M. Mortlock
Journal of Zoology (2020)
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
Download PDFEditorial Summary
A SARS-like virus in bats
Peter Daszak and colleagues identify two novel coronaviruses from Chinese horseshoe bats that are closely related to severe acute… show more- Sections
- Figures
- References
- Abstract
- Main
- Methods Summary
- Online Methods
- Accession codes
- References
- Acknowledgements
- Author information
- Ethics declarations
- Extended data figures and tables
- PowerPoint slides
- Rights and permissions
- About this article
- Further reading
- Comments
-
Bat Coronaviruses in China
settingsOpen AccessReview
by i Fan 1,2,Kai Zhao 1,2,Zheng-Li Shi 1,2
1
CAS Key Laboratory of Special Pathogens and Biosafety, Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan 430071, China
2
University of Chinese Academy of Sciences, Beijing 100049, China
*
Author to whom correspondence should be addressed.
Viruses 2019, 11(3), 210; https://doi.org/10.3390/v11030210
Received: 29 January 2019 / Revised: 26 February 2019 / Accepted: 26 February 2019 / Published: 2 March 2019
(This article belongs to the Special Issue Viruses and Bats 2019)
Download PDF
Browse Figures
Cite This Paper
Abstract
During the past two decades, three zoonotic coronaviruses have been identified as the cause of large-scale disease outbreaks–Severe Acute Respiratory Syndrome (SARS), Middle East Respiratory Syndrome (MERS), and Swine Acute Diarrhea Syndrome (SADS). SARS and MERS emerged in 2003 and 2012, respectively, and caused a worldwide pandemic that claimed thousands of human lives, while SADS struck the swine industry in 2017.
They have common characteristics, such as they are all highly pathogenic to humans or livestock, their agents originated from bats, and two of them originated in China. Thus, it is highly likely that future SARS- or MERS-like coronavirus outbreaks will originate from bats, and there is an increased probability that this will occur in China. Therefore, the investigation of bat coronaviruses becomes an urgent issue for the detection of early warning signs, which in turn minimizes the impact of such future outbreaks in China. The purpose of the review is to summarize the current knowledge on viral diversity, reservoir hosts, and the geographical distributions of bat coronaviruses in China, and eventually we aim to predict virus hotspots and their cross-species transmission potential.
Keywords: coronavirus; bat; epidemiology; cross-species; zoonosis
1. Introduction
Fifteen years after the first highly pathogenic human coronavirus caused the severe acute respiratory syndrome coronavirus (SARS-CoV) outbreak, another severe acute diarrhea syndrome coronavirus (SADS-CoV) devastated livestock production by causing fatal diseases in pigs. Both outbreaks began in China and were caused by coronaviruses of bat origin [1,2]. This increased the urgency to study bat coronaviruses in China to understand their potential of causing another virus outbreak.
In this review, we collected information from past epidemiology studies on bat coronaviruses in China, including the virus species identified, their host species, and their geographical distributions. We also discuss the future prospects of bat coronaviruses cross-species transmission and spread in China.2. Why Study Bat Coronaviruses in China?
2.1. Coronavirus Taxonomy
Coronaviruses (CoVs) belong to the subfamily Orthocoronavirinae in the family Coronaviridae and the order Nidovirales. CoVs have an enveloped, crown-like viral particle from which they were named after. The CoV genome is a positive-sense, single-strand RNA (+ssRNA), 27–32 kb in size, which is the second largest of all RNA virus genomes. Typically, two thirds of the genomic RNA encodes for two large overlapping polyproteins, ORF1a and ORF1b, that are processed into the viral polymerase (RdRp) and other nonstructural proteins involved in RNA synthesis or host response modulation. The other third of the genome encodes for four structural proteins (spike (S), envelope (E), mem..... https://www.mdpi.com/1999-4915/11/3/210/htm
https://www.mdpi.com/1999-4915/11/3/210/htm
Leave a comment:



Leave a comment: